Врожденные нарушения обмена витаминов
Утрата функции белка иногда вторична по отношению к сниженной доступности эссенциальной связанной молекулы, например кофактора фермента.
Дефицит витаминов в диете, например витамина В12 (кобаламина), вызывающий анемию и неврологическую симптоматику, и витамин D-дефицитный рахит иллюстрируют приобретенные нарушения этого типа. Унаследованные нарушения метаболизма витаминов также вызывают болезни.
Неудивительно, что фенотипы приобретенной и генетической болезни кофакторов часто перекрываются. Другими словами, приобретенная недостаточность витамина может быть частичной или полной фенокопией генетического заболевания.
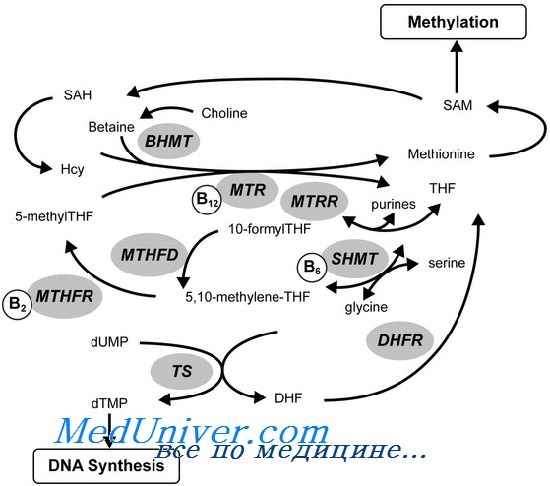
Например, вегетарианцы склонны к приобретенному недостатку витамина В12, и если у них развивается его недостаточность, они могут иметь биохимические аномалии, подобные тем, что имеются при гомоцистинурии, вызванной мутациями в ряде генов, нарушающими присоединение кофактора витамина В12, метилкобаламина, к ферменту метионинсинтетазе.
Метионинсинтетаза реметилирует гомоцистеин с образованием метионина, недостаточность метионинсинтетазы приводит к гомоцистинурии. Многочисленные наследственные нарушения транспорта или метаболизма витамина В12 (кобаламина) уменьшают доступность метилкобаламина и, следовательно, вторично нарушают активность метионинсинтетазы.
Несколько наследственных дефектов метаболизма витамина В12 снижают внутрикишечное всасывание кобаламина или транспорт его в другие клетки; другие нарушают специфический метаболизм кобаламина. Клинические проявления этих заболеваний разнообразны, но включают мегалобластную анемию и задержку физического развития. Эти аутосомно-рецессивные заболевания, как правило, частично или полностью лечатся высокими дозами витамина В12.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Врожденные нарушения обмена веществ
Опубликовано пн, 17/06/2019 — 19:08

Термин «врожденные ошибки метаболизма» ( “inborn errors of metabolism”) (IEM) был впервые предложен сэром Арчибальдом Гарродом ( Archibald Garrod) в 1908 году. Этим термином автор пытался описать те заболевания, которые вызваны блокированием метаболического пути из-за недостаточной активности конкретного фермента.
Диагноз врожденных нарушений обмена веществ (IEM) играет большую роль в педиатрии. Благодаря внедрению скрининга новорожденных (NBS), диагностика многих IEM стала относительно легкой с использованием лабораторных биомаркеров. Для большинства IEM ранняя диагностика предотвращает появление серьезных клинических симптомов, тем самым снижая заболеваемость и смертность. Однако из-за молекулярной, биохимической и клинической вариабельности IEM не все нарушения, включенные в программы NBS, будут обнаружены и диагностированы только скринингом.
В последние годы прогресс в таких технологиях, как тандемная масс-спектрометрия (МS / МS) и секвенирование следующего поколения (NGS), в которых используется стратегия массивного параллельного секвенирования, значительно расширили наши знания о IEM и метаболических нарушениях в целом. Эти новые технологии также позволили расширить и улучшить скрининг новорожденных (NBS) в глобальном масштабе. Приблизительно 80% расстройств, проверенных с помощью NBS являются IEM.
IEM можно классифицировать на две широкие категории: те, которые влияют на выработку энергии, и те, которые влияют на синтез или распад конкретных молекул или соединений. Хотя углеводы, жиры и белки используются в качестве источников энергии, степень использования определенного топлива зависит от типа органа или ткани. Нарушение метаболического процесса, который влияет на один тип энергии , приводит к увеличению использования альтернативного типа топлива; этот компенсаторный сдвиг в производстве энергии может привести к патологии аномальных метаболитов, наблюдаемой у пациентов с этими нарушениями, что является ключом к постановке диагноза. Благодаря комплексной регуляции обменных процессов во всем организме , накопление субстрата из заблокированной метаболической стадии будет задействовать альтернативные метаболические пути, которые обычно минимально используются в нормальных условиях, что приводит к увеличению производства и накопления потенциально вредных промежуточных метаболитов у пациентов с IEM. Эти промежуточные продукты могут также нарушать нормальный обмен веществ посредством активации или торможения ферментативных процессов или конкурентного действия, что приводит к появлению дополнительных клинических симптомов и патогномоничных паттернов повышенного уровня анализируемых веществ.
Большинство органических ацидурий, аминокислотных патологий, пероксисомных расстройств, лизосомальных нарушений накопления, нарушений накопления гликогена (GSD) и нарушений окисления митохондриальных жирных кислот являются примерами дефектных путей, при которых специфические ферменты расщепляют субстраты (гликоген, органические кислоты, аминокислоты или жирные кислоты) для производства энергии или для генерации основных строительных блоков, используемых в последующих процессах синтеза (например, синтез креатина). Напротив, порфирии, дефицит церебрального креатина и врожденные нарушения гликозилирования являются примерами дефектов в синтетических путях, которые влияют на выработку гема, креатина и гликопротеинов соответственно. Транспортеры и канальные белки, которые мобилизуют субстрат, попадают в обе категории.
Клинические фенотипы IEM являются широкими и часто неспецифичными, имитируя более распространенные состояния, причем, появление симптомов может проявиться в любом возрасте от плода до взрослого человека. Поскольку пациенты с более легкими мутациями и едва различимыми биохимическими фенотипами могут быть пропущены по предельным значениям, используемым для определения положительных результатов скрининга новорожденных , нормальный скрининг новорожденных не должен исключать эти расстройства из дифференциального диагноза у пациента, клиническая картина которого наводит на мысль о наследственном дефекте метаболизма. Клинические и биохимические фенотипы IEM у детей изучались десятилетиями; наоборот, варианты позднего или взрослого начала оставались в значительной степени нераспознанными. В последние годы взрослые формы многих наследственных метаболических нарушений все чаще идентифицируются как истинные легкие фенотипы, где симптомы в детстве не были достаточно серьезными, чтобы заслуживать изучения.
При оценке пациента на предмет возможной IEM обычные лабораторные анализы могут выявить основные паттерны, подозрительные для метаболического дефекта. Общие данные включают гипокетотическую гипогликемию, лактоацидоз, метаболический ацидоз, кетоз, гипераммонемию или метаболический ацидоз в сочетании с гипераммонемией. Оценка этих результатов анализа крови и мочи в сочетании с клинической картиной может сузить акцент на конкретном подмножестве метаболических нарушений. Среди подсказок, которые должны побудить клиницистов заподозрить IEM, есть такие сценарии, как больные новорожденные с ухудшением в анамнезе после неосложненной беременности, эпизодами болезни или колеблющимися симптомами летаргии или другими неврологическими симптомами, вызванными интеркуррентным заболеванием или стрессом, мультисистемным вовлечением, неспособность развиваться, задержка развития, прогрессирующие неврологические признаки или странные неврологические симптомы с или без психологических проблем у пациентов, у которых обычная этиология была исключена ( особенно у взрослых). Хотя биохимические генетические и молекулярно-генетические тесты необходимы для подтверждения диагноза, базовые лабораторные тесты все еще важны и часто дают первые ключи к возможно лежащему в основе IEM.
Первым шагом в выборе подходящего лабораторного исследования для исключения наследственногонарушения метаболизма является определение вероятности возникновения этого состояния из-за дефектов метаболизма малых молекул (таких как аминокислот, органических кислот, пуринов и пиримидинов, цикл мочевины, митохондриальный энергетический метаболизм) или нарушения метаболизма органелл (такие как лизосомы или пероксисомы). Пациенты с низкомолекулярными расстройствами обычно имеют острое начало заболевания, требующее экстренного вмешательства. Базовые лабораторные анализы должны проводиться у каждого ребенка с острым заболеванием, у которого возможно основное нарушение обмена веществ.
Пациенты с нарушениями метаболизма органелл обычно имеют неврологические и нервно-мышечные проявления, органомегалию, дисфункцию печени, с дисморфизмом или без него. Тем не менее, важно иметь в виду, что в некоторых случаях нарушения, влияющие на функцию органелл, могут также сопровождаться метаболическими кризисами, включая гипогликемию и / или метаболический ацидоз, требующие экстренного вмешательства. Кроме того, важно иметь ввиду , что некоторые IEM могут проявляться без метаболического кризиса, угрожающего жизни, причем , с такими необычными симптомами, как образование пузырей на коже после воздействия солнечного света и / или неврологические проявления (например, криптогенная боль в животе, парестезия или психотические эпизоды), как мы это видим в случаях большинства порфирий.
Метаболический ацидоз
Метаболический ацидоз — это нарушение кислотно-щелочного баланса в организме из-за потери бикарбоната, снижения почечной экскреции или увеличения выработки кислот. Определение кислотно-основного состояния важно при оценке состояния пациента с потенциальным наследственным метаболическим дефектом, поскольку высокий метаболический ацидоз с анионным зазором обычно вызывается накоплением органических кислот, включая молочную кислоту, кетоновые тела или необычные кислоты и их производные. Напротив, диарея и ацидоз почечных канальцев являются основными причинами метаболического ацидоза с нормальным разрывом анионов. При наличии ацидоза его следует оценивать в сочетании с другими метаболическими состояниями, такими как гипо- и гипергликемия, кетоз, гиперлактатемия и гипераммонемия.
Большинство IEM, которые представляют в клинической картине метаболический ацидоз и кетоз, являются органическими ацидемиями (то есть, метилмалоновая ацидемия, пропионовая ацидемия, изовалериановая ацидемия). С другой стороны, метаболический ацидоз с гипогликемией и отсутствием кетоза может быть единственной находкой лежащего в основе митохондриального дефекта окисления жирных кислот, где процесс борьбы с гипогликемией нарушен из-за неспособности вырабатывать энергию из метаболизма жирных кислот и увеличения производства -физиологические интермедиаты дикарбоновой кислоты. Метаболический ацидоз и гиперлактатемия в отсутствие повышенных органических кислот, кроме молочной и пировиноградной кислот, могут быть обнаружены при нарушениях метаболизма пировиноградной кислоты, а также при дефектах дыхательной цепи.
Нарушения углеводного обмена
Тяжелая гипогликемия представляет собой опасное для жизни состояние, встречающееся при многих нарушениях обмена веществ, в том числе нарушениях белкового обмена, таких как органическая ацидурия и некоторые аминокислотные патологии. Однако критическая гипогликемия является признаком, обнаруженным при нарушениях, непосредственно влияющих на углеводный обмен, таких как GSD, дефекты глюконеогенеза (дефицит глюкозо-6-фосфатазы, дефицит фруктозо-1,6-бифосфата) и дефекты окисления жирных кислот митохондриями, которые вызывают сильное истощение. циркулирующих и запасных углеводов, вторичных по отношению к дефектному производству альтернативной энергии.
При оценке гипогликемии логический оправданный подход заключается в том, чтобы сначала рассмотреть, является ли пациент «кетотическим» или «некетотическим». Нарушения митохондриального окисления жирных кислот, углеводного обмена, метаболизма кетоновых тел и органических ацидемий могут вызвать гипогликемию. Нарушения митохондриального окисления жирных кислот и кетогенеза, включая дефицит HMG-CoA-лиазы и дефицит HMG-CoA-синтазы, а также гиперинсулинемию, связаны с гипокетотической гипогликемией с или без выраженного метаболического ацидоза, тогда как другие нарушения, такие как органическая ацидемия, нарушение метаболизма тела кетонов и реже болезнь мочи кленового сиропа (MSUD), как правило, вызывает кетотическую гипогликемию.
В нормальных физиологических условиях, когда наступает гипогликемия, происходит одновременное превращение печеночного гликогена в глюкозу и увеличение катаболизма свободных жирных кислот. Дефекты окисления жирных кислот в митохондриях вызывают глубокую гипогликемию из-за истощения запасов глюкозы и гликогена в циркулирующей крови, возникающих из-за неспособности метаболизировать жирные кислоты для удовлетворения потребностей в энергии. При этих дефектах также существует неспособность преодолеть гипогликемию и снижение выработки ацетил-КоА из-за уменьшения потока через спираль бета-окисления, что влияет на выработку кетоновых тел.
При GSD наблюдается нарушение превращения печеночного гликогена в циркулирующую глюкозу во время голодания, что приводит к истощению доступных углеводов; гипогликемия связана с гепатомегалией, дисфункцией печени от легкой до тяжелой степени и гиперлактатемией. Тем не менее, гипогликемия может отсутствовать при GSD типа II (болезнь Помпе или дефицит лизосомной кислоты и мальтазы), поскольку цитоплазматический метаболизм гликогена сохраняется и гликоген накапливается только в лизосомах и на ранних стадиях GSD типа IV.
Гипогликемия также может быть обнаружена при нарушениях углеводного обмена, таких как галактоземия или наследственная непереносимость фруктозы. При классической галактоземии накопленный галактозо-1-фосфат ингибирует фосфоглюкомутазу, нарушая гликолиз, тогда как при наследственной непереносимости фруктозы накопленный фруктозо-1-фосфат ингибирует как глюконеогенез, так и гликогенолиз.
Гипогликемия в постпрандиальном состоянии или после непродолжительного голодания ( 8 часов) наводит на мысль о дефекте окисления жирных кислот. Гипогликемия после голодания средней продолжительности (4–8 часов) может быть вызвана гликогенозом или нарушением, влияющим на глюконеогенез. Другие основные лабораторные результаты также могут быть полезны; например, гипогликемия при наличии фиброза и цирроза печени может быть единственной находкой при наследственной тирозинемии I типа.
Гипераммонемия
Как и гипогликемия, гипераммонемия также угрожает жизни; следовательно, уровень аммиака в плазме должен быть проверен у всех пациентов с изменением сознания и энцефалопатией, особенно, у маленьких детей. Гипераммонемия может быть вызвана многими неметаболическими состояниями, включая заболевание печени, портокавальный шунт, гиперактивность глутаматдегидрогеназы или токсичность вальпроевой кислоты. Однако заметное повышение уровня аммиака, обычно в 10–100 раз превышающее верхний предел нормы, может быть связано с нарушениями цикла мочевины. Хотя некоторые органические ацидемии и нарушения митохондриального окисления жирных кислот также могут вызывать гипераммонемию, она обычно менее значительна.
Аммиак, нейротоксичный побочный продукт дезаминирования аминокислот, превращается в экскретируемую мочевину с помощью цикла мочевины в серии ферментативных стадий, происходящих либо в цитозоле, либо в митохондрии. Хотя цикл мочевины очень эффективен при нормальных условиях, он представляет собой сравнительно хрупкий метаболический процесс, на который могут влиять наследственные метаболические нарушения с помощью различных механизмов.
Нарушения цикла мочевины — это наследственные недостатки любого из ферментов цикла мочевины или продукции аллостерического кофактора N-ацетилглутамина, что приводит к тяжелой первичной гипераммонемии. Гипераммонемия также является относительно распространенной вторичной находкой в органических ацидуриях, где накопленные субстраты или промежуточные органические кислоты ингибируют фермент проксимальный цикл мочевины N-ацетилглутаматсинтазу (NAGS), вызывая общее снижение эффективности детоксикации цикла мочевины. Среди органических ацидурий пропионовая ацидемия и метилмалоновая ацидемия, в частности, могут проявляться перемежающейся вторичной гипераммонемией из-за ингибирующей способности накопленного пропионил-КоА. Метаболические нарушения, при которых циркулирует уровень орнитина, цитруллина, или аргинин снижается из-за почечных потерь или снижение эндогенной продукции также может вызывать гипераммонемию, поскольку все три являются промежуточными циклами мочевины. Когда циркулирующие или внутриклеточные уровни этих аминокислот падают, эффективность цикла мочевины может снижаться, что приводит к гипераммонемии; наиболее глубокие потери аминокислот, важных для цикла мочевины, обнаруживаются при цистинурии и непереносимости лизинурического белка, когда почечная реабсорбция орнитина и аргинина может быть значительно снижена из-за конкуренции за общий переносчик двухосновных аминокислот. Напротив, нарушения окисления митохондриальных жирных кислот могут сопровождаться гипераммонемией вследствие комбинированных эффектов истощения субстрата и ингибирования цикла мочевины токсичными видами ацилкарнитина. Уровень ацетил-КоА, конечного продукта бета-окисления жирных кислот, уменьшается, когда общий поток через путь уменьшается. Ацетил-КоА необходим для производства N-ацетилглутамата, который аллостерически активирует фермент карбамоилфосфат-синтетазу 1 (CPS1); CPS1 преобразует аммиак в карбамоилфосфат на ограничивающей скорость первой стадии цикла мочевины. При некоторых дефектах окисления жирных кислот с длинной цепью ацилирование жирных остатков активного сайта CPS1 непосредственно влияет на способность детоксикации цикла мочевины. Наконец, некоторые расстройства могут вызывать гипераммонемию, вторичную по отношению к повреждению органов. Например, при лизосомных расстройствах, включая мукополисахаридозы, а также при некоторых пероксисомных расстройствах, накопление и хранение сложных крупных молекул в печени приводит к гепатоцеллюлярному повреждению, что, в свою очередь, вызывает снижение эффективности цикла мочевины.
Лактоацидоз
Физиологический баланс циркулирующей молочной кислоты, поддерживаемый продукцией молочной кислоты посредством цитоплазматического гликолиза и многокомпонентного митохондриального потребления, может быть нарушен как неметаболическими, так и метаболическими факторами . Лактоацидоз может возникать из-за увеличения выработки лактата или снижения его метаболизма. У большинства метаболических расстройств с гиперлактатемией наблюдается параллельный кетоз, за исключением дефицита пировиноградной дегидрогеназы, гликогеноза I типа или некоторых нарушений окисления жирных кислот. Лактоацидоз чаще всего вызывается гипоксией тканей из-за плохой циркуляции или недостаточного снабжения кислородом по нескольким причинам, включая кардиогенный или гиповолемический шок. Неметаболические источники гиперлактатемии обычно не сопровождаются кетозом. Однако некоторые IEM, включая органические ацидурии, нарушения метаболизма митохондриальной энергии или дефекты глюконеогенеза, также могут проявляться лактоацидозом.
Как только неметаболическая этиология гиперлактатемии была исключена, наиболее распространенными причинами гиперлактатемии, вторичной по отношению к нарушению митохондриальной энергии токсическими метаболитами, являются нарушения окисления жирных кислот, органические ацидурии и, в очень редких случаях, нарушения цикла мочевины. Другие наследственные причины персистирующей гиперлактатемии включают нарушения метаболизма гликогена, нарушения, влияющие на глюконеогенез, и расстройства, непосредственно влияющие на цикл Кребса или метаболизм пировиноградной кислоты. При дефектах глюконеогенеза, таких как дефицит фруктозы-1, 6-фосфатазы и GSD типа IA [дефицит глюкозо-6-фосфатазы (G-6PD)], пики гиперлактатемии отмечаются при голодании или в гипогликемических ситуациях. . При нарушениях, влияющих на деградацию гликогена, гиперлактатемия достигает пика после приема пищи. Гиперлактатемия может различаться при нарушениях, непосредственно влияющих на метаболизм пировиноградной кислоты. При дефиците пируватдегидрогеназы, дефиците альфа-кетоглутаратдегидрогеназы и нарушениях дыхательной цепи гиперлактатемия обычно возникает в состоянии сытости, тогда как при дефиците пируваткарбоксилазы гиперлактатемия возникает как в состоянии натощак, так и в состоянии сытости. При оценке пациента на предмет гиперлактатемии часто упускают из виду соотношение между молочная кислота / пировиноградная кислота и 3-ОН масляная кислота / ацетоуксусная кислота. Молярное соотношение молочной кислоты / пировиноградной кислоты в плазме коррелирует с отношением NAD + / NADH и может косвенно отражать цитозольное окислительно-восстановительное состояние, тогда как соотношение 3-гидроксибутирная кислота / ацетоуксусная кислота отражает внутримитохондриальное окислительно-восстановительное состояние.
Кетонурия
Кетоновые тела 3-гидрокси-масляная кислота, ацетоуксусная кислота и ацетон являются естественными конечными продуктами бета-окисления митохондриальных жирных кислот. Кетонурия, увеличение экскреции кетонов с мочой, обнаруживается физиологически в позднем младенчестве, детстве и подростковом возрасте, но не считается нормальным у новорожденных. Физиологический кетоз не сопровождается метаболическим ацидозом, гиперлактатемией или гипогликемией — маркерами метаболического стресса. Это обычное явление после голодания, рвоты, употребления кетогенной диеты или повышенного уровня катаболизма. Однако, поскольку кетоны являются органическими кислотами, тяжелая кетонурия, которая вызывает метаболический ацидоз, не должна рассматриваться как физиологическая и указывать на врожденные нарушения метаболизма. Время кетонурии в связи с кормлением или голоданием является важным фактором, который может указывать на потенциальный тип основного метаболического расстройства. Например, тяжелая кетонурия с гипогликемией натощак или постпрандиальная гипергликемия с гиперлактатемией являются общими признаками при некоторых нарушениях гликогеноза.
В отличие от других маркеров метаболического стресса, кетоз является клинически значимым как при повышении, так и при его отсутствии. В то время как серьезное снижение экскреции кетонов наряду с низким уровнем циркулирующей глюкозы (гипокетотическая гипогликемия) является обычным явлением при рвоте, анорексии или генерализованных катаболических состояниях, этот паттерн также является существенным индикатором потенциального нарушения окисления митохондриальных жирных кислот, с или без чрезмерного использования глюкозы. Однако важно отметить, что некоторые нарушения окисления жирных кислот в митохондриях могут сопровождаться периодическими эпизодами кетонурии от легкой до тяжелой степени, когда пораженный фермент достаточно дистален в пути деградации бета-окисления, что метаболизм длинноцепочечных жирных кислот все еще способен генерирование некоторых кетонов, как в случае дефицита 3-гидроксиацил-СоА-дегидрогеназы (HAD) или дефицита со средней цепью ацил-КоА-дегидрогеназы (MCAD).
Нейровизуализация
Помимо исследований, перечисленных выше, другие важные начальные тесты включают CBC, функциональные тесты печени, исследования коагуляции, уровни креатинкиназы, тесты почечной функции, BUN, исследование мочевой кислоты, липидных профилей и клеток CSF и глюкозу CSF.
Исследования нейровизуализации, включая МРТ и МР-спектроскопию, могут показать специфические изменения мозга, характерные для определенных наследственных дефектов, таких как симметричные базальные ганглии и вовлечение таламуса, наблюдаемые при митохондриальных расстройствах, субдуральных выпотах и сниженной оперкуляризации, наблюдаемой при глютариновой ацидемии типа I (GA1), отсутствующий пик креатина МР спектроскопия демонстрирует врожденные нарушения метаболизма креатина и изменения белого вещества при таких нарушениях, как Х-сцепленная адренолейкодистрофия и метахроматическая лейкодистрофия.
Лабораторные тесты
Лабораторные тесты, которые представляют широкую сеть и могут использоваться для диагностики множественных IEM, включают анализ мочи на органическую кислоту, и аминокислоты в плазме и моче, ацилкарнитин в плазме и анализ жирных кислот с очень длинной цепью в сыворотке. Поскольку вторичный дефицит карнитина, являющийся следствием потери карнитина в виде сложных эфиров ацилкарнитина в моче, является распространенным явлением при многих нарушениях обмена веществ, включая нарушения окисления жирных кислот и органические ацидурии, при оценке состояния пациента на предмет потенциального нарушения обмена веществ следует также включать количественное определение содержания карнитина в сыворотке.
Некоторые расстройства приводят к увеличению циркулирующих промежуточных метаболитов, которые, благодаря своему характеру, образуют основу для диагностики и диетического мониторинга (т.е. повышенный уровень фенилаланина в плазме и пониженный уровень тирозина при фенилкетонурии, повышенное содержание свободных жирных кислот при расстройствах окисления жирных кислот или повышенное число разветвленных цепей). аминокислоты при MSUD); другие расстройства приводят к повышенной экскреции ключевых метаболитов, которые могут использоваться для тех же целей (например, повышенный цистин мочи при цистинурии), в то время как некоторые расстройства вызывают несколько изменений. Увеличение содержания аномальных метаболитов в плазме также могут быть обнаружены и в моче, если концентрация крови достигает почечного порога для того или иного аналита. В целом, нарушения метаболизма аминокислот, которые влияют на стадии проксимального метаболизма (например, фенилкетонурия или MSUD), приводят к аномальным аминокислотным профилям плазмы.
Нарушения метаболизма аминокислот, которые влияют на дистальные ферментативные стадии (то есть изовалериановая ацидемия или глутаровая ацидурия типа I), могут или не могут иметь отклонения в анализе аминокислот в плазме, но, как правило, приводят к аномальным профилям мочевой органической кислоты. Нарушения, которые влияют на переносчики аминокислот в почках (например, цистинурия или непереносимость белка лизинурия), обычно лучше всего диагностируются по аминокислотному профилю мочи.
Состояния, влияющие на различные стадии общего метаболического пути, могут приводить к аномальным паттернам общих промежуточных метаболитов; в таких случаях может потребоваться дальнейшее тестирование, чтобы сузить диагноз, например, когда в плазменном профиле ацилкарнитина обнаружен повышенный уровень 3-гидроксиизовалерил- / 2-метил-3-гидроксибутирилкарнитина (C5OH). Этот аналит обнаружен при нескольких нарушениях, влияющих на метаболизм аминокислот с разветвленной цепью. В таких случаях может потребоваться анализ мочи на органическую кислоту или дополнительные биохимические или молекулярные анализы для подтверждения первоначальных результатов или для постановки окончательного диагноза.
В некоторых других условиях анализ активности фермента и / или молекулярный анализ могут быть необходимы для конкретного диагноза; следует отметить, что некоторые ферментные тесты требуют инвазивных процедур, таких как биопсия печени при заболевании хранения гликогена в печени или биопсия кожи при культивировании фибробластов для исследований окисления жирных кислот.
Источник