Наследственные болезни обмена веществ, проявляющиеся в периоде новорожденности
Е.Д. Белоусова, М.Ю. Никанорова, Е.А. Николаева
Московский НИИ педиатрии и детской хирургии Минздрава РФ
Статья посвящена актуальной и малоизвестной широкому кругу врачей проблеме — диагностике и лечению врожденных дефектов метаболизма в неонатальном периоде. Описываются симптомы, на основании которых педиатр и невролог могут заподозрить у новорожденного метаболическое заболевание, приводится алгоритм необходимых лабораторных исследований. Кратко изложены общие принципы лечения неонатальных врожденных дефектов метаболизма.
Диагностика и лечение врожденных аномалий метаболизма — одна из самых сложных задач как для невролога, так и для педиатра. С одной стороны, трудности диагностики связаны с клиническим полиморфизмом заболеваний, по-разному протекающих у детей различного возраста, с другой — с тем, что самые различные нарушения обмена в одной и той же возрастной группе могут иметь сходные клинические проявления [1].
Особые трудности вызывает диагностика врожденных дефектов метаболизма у новорожденного [2]. Заболевания протекают тяжело и заканчиваются летально до того, как становится очевидной специфическая метаболическая и неврологическая картина. В периоде новорожденности проявляются примерно 25% известных наследственных болезней обмена веществ и почти все они отличаются особой тяжестью состояния ребенка. По остроте начала и течению наследственные болезни обмена веществ в неонатальном периоде могут напоминать нейроинфекции и нейродистресс-синдром. Однако явная прогредиентность течения с утратой функций не характерна для новорожденного, так как сами эти функции еще не сформированы. Поэтому большинство детей с неонатальными формами наследственных дефектов метаболизма либо умирают в первые месяцы жизни без установленного диагноза, либо длительно наблюдаются с самыми разными неадекватными диагнозами. Чаще всего наследственные болезни обмена скрываются под маской детского церебрального паралича, умственной отсталости и эпилепсии, резистентной к антиконвульсантам. Причины неонатальных наследственных метаболических нарушений многочисленны (см. ниже).
Этиология наследственных заболеваний обмена веществ с манифестацией клинических проявлений в неонатальном периоде
Нарушения метаболизма аминокислот
Нарушения цикла синтеза мочевины (дефицит N-ацетилглутаматсинтетазы, карбамоилфосфатсинтетазы, орнитинкарбамоилтрансферазы, аргининсукцинатсинтетазы, аргининсукцинатлиазы и аргиназы)
Гиперорнитинемия — гипераммониемия — гомоцитруллинурия
Болезнь с запахом мочи кленового сиропа (лейциноз)
Нарушения метаболизма органических кислот
Нарушения в системе пируватдегидрогеназного комплекса (дефицит пируваткарбоксилазы, пируватдегидрогеназы, дигидролипоилтрансацетилазы и дигидролипоилдегидрогеназы) Нарушения функций дыхательной цепи (дефициты ферментов I-IV комплекса дыхательной цепи) Нарушения функций ферментов цикла Кребса (дефицит фумаразы, сукцинатдегидрогеназы, $\alpha$-кетоглутаратдегидрогеназы и аконитазы) Нарушения синтеза и обмена карнитина (системная недостаточность карнитина, дефицит карнитинпальмитоилтрансферазы II и ацилкарнитинтранслоказы) Нарушения $\beta$-окисления жирных кислот (дефициты ацил-СоА-дегидрогеназ жирных кислот с короткой, средней, длинной и очень длинной углеродной цепью, множественный дефицит ацил-СоА-дегидрогеназ)
Нарушения обмена биотина
Дефицит синтетазы голокарбоксилаз
Дефицит ацил-СоА-оксидазы пероксисом
Дефицит 3-оксиацил-СоА-тиолазы пероксисом
Бифункциональная недостаточность протеинов
Лейкодистрофия Канавана — Ван Богарта — Бертранда
Лейкодистрофия Пелицеуса — Мерцбахера
Нарушения обмена углеводов
Наследствення непереносимость фруктозы
Галактоземия (дефицит галактозо-1-фосфатуридилтрансферазы)
Редко, но встречаются неонатальные формы таких заболеваний, как болезнь Менкеса, гликогеноз типа II, болезнь Нимана-Пика и Гоше, синдром Барта, тирозинемия I типа, болезнь Альперса, а также сиалидоз [3].
Эпидемиология. Данные о распространенности наиболее изученных наследственных метаболических заболеваний приведены в табл. 1.
| Таблица 1. Распространенность наследственных заболеваний обмена веществ с неонатальным дебютом | |
| Заболевание | Частота |
| Дефицит аргининсукцинатлиазы | 1/70 000 |
| Дефицит карбамоилфосфатсинтетазы | 1/1 000 000-1/800 000 |
| Дефицит аргининсукцинатсинтетазы | 1/250 000 |
| Некетотическая гиперглицинемия | 1/55 000 |
| Болезнь с запахом мочи кленового сиропа | 1/200 000 |
| Изовалериановая ацидемия | 1/200 000 |
| Пропионовая ацидемия | 1/350 000 |
| Метилмалоновая ацидемия | 1/48 000 |
| Дефицит ацил-СоА-дегидрогеназы жирных кислот со средней длиной углеродной цепи | 1/9 000-1/13 000 |
| Синдром Цельвегера | 1/50 000-1/100 000 |
| Дефицит биотинидазы | 1/35 000-1/54 000 |
| Дефицит орнитинкарбамоилтрансферазы | 1/80 000 |
| Дефицит аргининсукцинатлиазы | 1/90 000 |
| Тирозинемия I типа | 1/30 000 |
| Аргининемия | 1/990 000 |
Распространенность других, менее изученных дефектов метаболизма неизвестна. Число описаний больных с редкими неонатальными метаболическими заболеваниями колеблется от единичных до сотен наблюдений.
Генетические данные. Большинство врожденных дефектов метаболизма с проявлениями в неонатальном периоде имеют аутосомно-рецессивный тип наследования. Исключениями из правила являются неонатальная адренолейкодистрофия, лейкодистрофия Пелицеуса-Мерцбахера, болезнь Менкеса и синдром Барта, которые наследуются по рецессивному сцепленному с Х-хромосомой типу, и дефицит орнитинкарбамоилтрансферазы, который передается доминантно, в сцеплении с Х-хромосомой. Дефицит пируватдегидрогеназы и множественный дефицит ацил-СоА-дегидрогеназ имеют два возможных типа наследования — аутосомно-рецессивный и рецессивный, сцепленный с Х-хромосомой. Нарушения в системе транспортной цепи электронов в дыхательной цепи наследуются по митохондриальному типу (материнское наследование). Дефицит I комплекса дыхательной цепи (NADH; CoQ-редуктазы) наследуется как по аутосомно-рецессивному, так и по митохондриальному типу, описаны также X-cцепленные формы.
Значительные успехи достигнуты в молекулярной диагностике неонатальных дефектов метаболизма. В табл. 2 представлены врожденные метаболические заболевания с картированными генами.
| Таблица 2. Локализация генов, ответственных за развитие наследственных заболеваний обмена веществ с неонатальным дебютом | |
| Заболевание | Хромосома, локус |
| Болезнь с запахом мочи кленового сиропа: | |
-ген Е1 | 19q13.1-q13.2 |
-ген Е1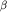 | 6p22-p21 |
| -ген Е2 | 1p31 |
| -ген Е3 | 7 |
| Изовалериановая ацидемия | 15q13-q15 |
| Пропионовая ацидемия: | |
— -cубъединица -cубъединица | 13q32 |
| —-субъединица | 3q21-q22 |
| Метилмалоновая ацидемия | 6р21.2-р12 |
| Дефицит пируваткарбоксилазы | 11 (длинное плечо) |
| Дефицит пируватдегидрогеназы: | |
| —-cубъединица | Хр22-сеп |
| —-субъединица | 3 |
| Дефицит дегидролипоилдегидрогеназы | 7q31-q32 |
| Дефицит IV комплекса дыхательной цепи: | |
| -4-я субъединица | 16q22 |
| -Vb-cубъединица | 2сеп-q13 |
| Дефицит фумаразы | 1q42.1 |
| Дефицит сукцинатдегидрогеназы | 1р35-р36.1 |
| Дефицит карнитинпальмитоилтрансферазы II | 1р11-р13 |
| Синдром Барта | Хq28 |
| Болезнь Менкеса | Хq13.3 |
| Некетотическая гиперглицинемия | 9 (короткое плечо) |
| Дефицит ацил-СоА-дегидрогеназы жирных кислот: | |
| -со средней длиной углеродной цепи | 1р22.1 |
| -с короткой углеродной цепью | 12 |
| -с длинной углеродной цепью | 2q34-q35 |
| Дефицит биотинидазы | 3р25 |
| Дефицит синтетазы голокарбоксилаз | 21q22.1 |
| Синдром Цельвегера | 8q21 |
| Болезнь Kраббе | 14q21-q31 |
| Лейкодистрофия Kанавана — Ван Богарта — Бертранда | 17р13-pter |
| Метахроматическая лейкодистрофия | 22q13.31-qter |
| Лейкодистрофия Пелицеуса-Мерцбахера | Хq22 |
| Множественный дефицит ацил-СоА-дегидрогеназ (дефицит электроннотранспортного флавопротеина) | 15q23-25 |
Общая клиническая характеристика врожденных дефектов метаболизма в неонатальном периоде
Несмотря на то, что точный диагноз возможен только при применении лабораторных методов исследования, важное значение имеют анамнестические и клинические данные, на основании которых врач может заподозрить наличие у новорожденного врожденного дефекта метаболизма.
Анамнез. На наследственное заболевание метаболизма указывают сведения о возможном близкородственном браке родителей, неврологическая симптоматика у одного из родителей, наличие сибса, страдающего неврологическим заболеванием или умершего по неясной причине в неонатальном периоде, младенчестве или детстве. Как правило, тяжесть состояния самого новорожденного не соответствует относительно благополучному течению данной беременности и родов, т. е. отсутствуют указания в анамнезе на тяжелую гипоксию плода и асфиксию новорожденного, травматические повреждения нервной системы.
Начало заболевания. Для многих неонатальных нейрометаболических заболеваний характерен продолжительностью в несколько дней от рождения до первых клинических проявлений заболевания. является хоть и частым, но не обязательным симптомом при наследственных болезнях обмена веществ.
Нарушения режима сна — бодрствования. Для здорового доношенного новорожденного характерны периоды спонтанно возникающего бодрствования, когда ребенок просыпается, двигается и плачет. Для недоношенного со сроком гестации от 30 до 34 нед характерно в основном стимулированное бодрствование (при стимуляции ребенок просыпается, двигается и плачет), имеется также кратковременное спонтанное бодрствование. У новорожденного с наследственным заболеванием обмена веществ возможны все степени тяжести нарушения уровня бодрствования (нарушения сознания) — от легкой сонливости до тяжелой комы.
Симптомокомплекс является типичным для наследственных аномалий метаболизма. Этим термином обозначается генерализованная мышечная гипотония. Симптомокомплекс включает позу , снижение сопротивления к пассивным движениям, увеличение объема движений в суставах, снижение общей двигательной активности и задержку моторного развития. Мышечная гипотония и снижение сухожильных рефлексов, характерные для наследственных дефектов метаболизма, позднее (на 1-2-м году жизни) сменяются мышечной дистонией и спастичностью. Исключением являются неонатальная форма лейкодистрофии Краббе и болезнь с запахом мочи кленового сиропа, при которых уже в неонатальном периоде возможно повышение мышечного тонуса.
Неонатальные судороги — один из наиболее частых симптомов врожденных дефектов метаболизма. Наиболее часто — это миоклонии и минимальные судорожные проявления (непроизвольные движения в области лица или конечностей: насильственное открывание глаз или моргание, тоническая девиация глазных яблок, жевательные и глотательные движения, сосание и др.). Как правило, судороги метаболического генеза повторяются и плохо поддаются лечению антиконвульсантами. Возможен эпилептический статус.
Необъяснимые трудности вскармливания. Для наследственных дефектов метаболизма характерны нарушения сосания и глотания. В тяжелых случаях ребенок не сосет грудь и не глотает, что требует зондового кормления. В менее тяжелых случаях сосание нарушено, новорожденный неплотно обхватывает губами сосок, отмечается постоянное подтекание молока из углов рта и поперхивания при глотании. Нарушения сосания и глотания при врожденных дефектах обмена могут иметь различное происхождение: дисплазия двигательных зон коры головного мозга, отвечающих за орофарингиальную мускулатуру, за счет пренатального воздействия токсичных метаболитов у плода, летаргия у новорожденного, общая мышечная гипотония, которая захватывает и мышцы, отвечающие за сосание и глотание. В дальнейшем у больных отмечаются нарушения жевания и дизартрия, так как данные функции обеспечиваются той же мускулатурой.
Дыхательные нарушения нередко встречаются при наследственных заболеваниях обмена веществ. Гиперпноэ и апноэ наблюдаются при отсутствии заболеваний сердца или легких и респираторного дистресс-синдрома. Их возникновение связано с воздействием токсичных продуктов метаболизма на дыхательный центр или сопутствующим метаболическим ацидозом.
Увеличение размеров печени и селезенки в сочетании с неврологической симптоматикой у новорожденного с высокой вероятностью свидетельствует о возможном наличии наследственного заболевания обмена веществ, хотя также может отмечаться при инфекционных и гематологических болезнях. Увеличение размеров печени характерно для болезней пероксисом, митохондриальных энцефаломиопатий, нарушений обмена аминокислот и органических кислот, а также для более редких неонатальных форм болезней накопления (лейкодистрофии Краббе, болезней Нимана — Пика и Гоше, сиалидоза и др.).
Анорексия и рвота также относятся к общим симптомам, наблюдающимся при многих врожденных метаболических заболеваниях. Они особенно характерны для гипераммониемий, органических ацидемий и нарушений -окисления жирных кислот. Более редким симптомом поражения желудочно-кишечного тракта является диарея. Как правило, все эти симптомы вызывают задержку прибавки массы у новорожденного, и их легко принять за желудочно-кишечные заболевания.
Помимо общих клинических проявлений, характерным симптомом многих метаболических заболеваний является черепно-лицевая дисморфия (табл. 3).
| Таблица 3. Черепно-лицевая дисморфия при различных наследственных заболеваниях обмена веществ с неонатальным дебютом | |
| Заболевание | Черепно-лицевая дисморфия |
| Синдром Цельвегера и другие болезни пероксисом | Брахицефалия, одутловатое, плоское лицо с опухшими веками, высокий лоб, сглаженные надбровные дуги, гипертелоризм, эпикант, монголоидный разрез глаз, микрогнатия * |
| Множественный дефицит ацил-СоА-дегидрогеназ | Высокий лоб, низко расположенные ушные раковины, гипертелоризм |
| Дефицит пируватдегидрогеназы | Вздернутый нос с широкой спинкой, низко посаженные ротированные кзади ушные раковины, микрогнатия |
| Дефицит кофактора молибдена | Треугольное лицо, курносый нос с широкой переносицей, энофтальм, телекант, расщелина мягкого неба |
| Мевалоновая ацидурия | Широкая переносица, микрогнатия, низко расположенные ушные раковины |
| * При других болезнях пероксисом (кроме болезни Цельвегера) черепно-лицевая дисморфия менее постоянна и менее выражена. | |
Характерные для отдельных наследственных заболеваний обмена веществ изменения кожи, внутренних органов и глаз приведены в табл. 4.
| Таблица 4. Экстраневрологические проявления наследственных врожденных дефектов метаболизма в неонатальном периоде | ||
| Объект поражения | Характер поражения | Заболевания |
| Kожа | Эритематозные высыпания | Пропионовая ацидемияМетилмалоновая ацидемия Дефицит синтетазы голокарбоксилаз Болезнь с запахом мочи кленового сиропа Тирозинемия I типа Дефицит ацил-СоА-дегидрогеназы жирных кислот с короткой углеродной цепью |
| Глаза | Kатаракта | Синдром Цельвегера и другие болезни пероксисомГалактоземия Мевалоновая ацидурия |
| Пигментная дегенерация сетчатки | Синдром Цельвегера и другие болезни пероксисомДефицит биотинидазы | |
| Дисплазия сетчатки | Дефицит сульфитоксидазы и кофактора молибденаДефицит биотинидазы | |
| Дислокация хрусталика. Глаукома | Синдром Цельвегера и другие болезни пероксисом | |
| Сердце | Kардиомиопатия | Дефицит I и IV комплексов дыхательной цепиНарушение -окисления митохондриальных жирных кислот Дефицит карнитинпальмитоилтрансферазы II Синдром Барта |
| Почки | Поликистоз | Синдром Цельвегера и другие болезни пероксисомДефицит карнитинпальмитоилтрансферазы II Множественный дефицит ацил-СоА-дегидрогеназ |
| Поликистоз + диспластические изменения. Синдром Де Тони — Дебре — Фанкони | Дефицит III и IV комплексов дыхательной цепи | |
| Поджелудочная железа | Панкреатит | Изовалериановая ацидемияМетилмалоновая ацидурия Болезнь с запахом мочи кленового сиропа |
Специфический запах мочи является важным диагностическим признаком и встречается при лейцинозе (запах или сена), изовалериановой ацидемии и множественном дефиците ацил-СоА-дегидрогеназ (и при том, и при другом заболевании характерен запах или ), дефиците биотинидазы, дефиците синтетазы голокарбоксилаз, -метилкротонилглицинурии (запах ), тирозинемии I типа (запах ) и при болезни с запахом мочи кленового сиропа [5].
Клиническая диагностика. В типичном случае наследственного дефекта метаболизма новорожденный рождается доношенным от физиологически протекающих родов, и его состояние после рождения вполне благополучно. Через некоторое время (1-3 дня) состояние ребенка ухудшается, развиваются летаргия или ступор, появляются судороги, дыхательные расстройства, анорексия и рвота. В зарубежной литературе такое состояние получило наименование . Для неврологического статуса характерны диффузная мышечная гипотония и нарушения сосания и глотания. Подозрение на наличие врожденного дефекта метаболизма получает подтверждение, если имеется отягощенность родословной по неврологическим заболеваниям, специфический запах мочи и экстраневрологические проявления — кардиомиопатия, гепатоспленомегалия и др.
Лабораторная диагностика наследственных дефектов метаболизма в неонатальном периоде
При подозрении на наследственное нарушение обмена веществ рекомендуется на первом этапе проведение стандартных клинических и биохимических исследований крови. О возможном наличии метаболического заболевания свидетельствуют:
— анемия, лейко- и тромбоцитопения (характерны для изовалериановой, пропионовой и метилмалоновой ацидемий);
— нейтропения (характерна для синдрома Барта);
— ацидоз в сочетании с кетозом (характерен для большинства органических ацидемий);
— повышение содержания лактата в крови (характерно для нарушений метаболизма пирувата, дефектов дыхательных комплексов и ферментов -окисления жирных кислот);
— гипогликемия с низким содержанием кетоновых тел (характерна для нарушений митохондриального -окисления жирных кислот);
— гипераммониемия (характерна для нарушений цикла мочевины, дефицита ацил-СоА-дегидрогеназ жирных кислот со средней длиной углеродной цепи и для большинства органических ацидемий).
На втором этапе обследования при наличии хотя бы одного из перечисленных выше лабораторных нарушений показано более детальное лабораторное обследование новорожденного, включающее определение спектра аминокислот крови и мочи, органических кислот в моче, карнитина и ацилкарнитинов в крови (см. таб. 5).
| Таблица 5. Алгоритм лабораторной диагностики при предполагаемом наследственном дефекте метаболизма у новорожденного | ||
| Гипераммониемия + дыхательный алкалоз | Определение содержания аминокислот в крови и моче | Болезни цикла биосинтеза мочевины |
| Kетоацидоз + гипогликемия | Определение содержания аминокислот в крови и моче | Органические ацидемии |
| Определение содержания органических кислот в моче | ||
| Определение содержания глюкозы, галактозы, редуцирующих веществ в моче | Наследственные нарушения обмена углеводов | |
| Гипокетонемия + гипогликемия | Определение содержания органических кислот в моче | Болезни транспорта и -окисления жирных кислот |
| Определение содержания ацилкарнитина в моче | ||
| Определение уровня карнитина в крови | ||
| АминоацидурияАминоацидемия | Определение содержания аминокислот в крови и моче | Аминоацидопатии |
| Лактат-ацидоз | Определение уровня лактата, пирувата, кетоновых тел в крови | Митохондриальные энцефалопатии |
| Гистохимическое определение активности митохондриальных ферментов в биоптате мышцы | ||
| Определение уровня жирных кислот с очень длинной углеродной цепью в крови | Пероксисомные болезни | |
| Определение уровня фитановой и пипеколовой кислот в крови и моче | ||
Следующим этапом диагностики является определение активности ферментов, дефицитом которых вызвано заболевание, в культуре фибробластов, лимфоцитах или биоптатах мышцы и печени. На завершающем этапе проводится молекулярная диагностика патологии с помощью ДНК-зондов.
Нейросонографические исследования в неонатальном периоде, как правило, малоспецифичны и более важны для дифференциального диагноза с гипоксически-ишемическими поражениями и внутричерепными кровоизлияниями у новорожденных [6]. Возможно наличие пренатальных поражений коры и белого вещества, базальных ядер и мозолистого тела в виде снижения плотности указанных образований. Более информативной является магнитно-резонансная томография головного мозга, которая проводится на 1-м году жизни ребенка и может демонстрировать кортикальные дисгенезии, гипоплазию мозолистого тела и мозжечка, а также повреждения белого вещества головного мозга.
Результаты электроэнцефалографии, электронейромиографии, исследования вызванных корковых потенциалов в неонатальном периоде неспецифичны и не могут использоваться для дифференциальной диагностики.
Дифференциальный диагноз. Сходные с наследственными дефектами метаболизма клинические симптомы (неврологические симптомы, нарушения дыхания и сознания, судороги, мышечная гипотония, рвота) часто встречаются при гипоксически-ишемических энцефалопатиях, пороках развития головного мозга, внутриутробных инфекциях или внутричерепных кровоизлияниях. Основные анамнестические и клинические различия наследственных болезней обмена и гипоксически-ишемической энцефалопатии новорожденных приведены в табл. 6.
| Таблица 6. Дифференциальный диагноз наследственных заболеваний обмена веществ и гипоксически-ишемической энцефалопатии в неонатальном периоде | ||
| Симптом | Гипоксически-ишемическая энцефалопатия | Наследственная болезнь метаболизма |
| Отягощенность родословной по неврологическим заболеваниям | — | + |
| Гипоксия плода | + | — |
| Отягощенное течение беременности и родов | + | — |
| Заболевания матери, вызывающие нарушения плацентарного кровотока (хронические воспалительные заболевания половой сферы, патология плаценты, артериальная гипертензия и др.) | + | — |
| Мекониальное окрашивание околоплодных вод | + | — |
| Недоношенность | + | * |
| Появление неврологических нарушений сразу после рождения | + | — |
| Черепно-лицевая дисморфия | — | + |
| Гепатомегалия, кардиомиопатия, поликистоз почек | — | + |
| Нейросонографические изменения, характерные для гипоксически-ишемической энцефалопатии | + | — |
| Примечание. * возможна, но не характерна. | ||
Лечение. Во многих случаях необходимы экстренные терапевтические мероприятия, направленные на борьбу с жизнеугрожающими ацидозом, гипогликемией, гипераммониемией и гиповолемией [7]. Концентрация токсичных метаболитов в крови должна быть нормализована как можно быстрее, чтобы предотвратить необратимое повреждение головного мозга. Наиболее эффективным способом лечения в остром периоде является гемодиализ.
Принципы лечения генетически детерминированных дефектов метаболизма можно условно разделить на и [2]. К подходам относят замену дефектного фермента или гена, ответственного за синтез фермента. Данный подход обеспечивает кардинальное решение проблем, связанных с метаболическими расстройствами. Однако, к сожалению, вмешательство в настоящее время невозможно.
К подходам к терапии относятся:
— уменьшение поступления в организм веществ, обмен которых нарушен;
— введение недостающего метаболита;
— удаление токсичных веществ;
— сведение к минимуму введения препаратов или действия других факторов, способствующих ухудшению состояния больного;
— назначение витаминов, являющихся кофакторами дефектных ферментов;
— уменьшение образования токсичных метаболитов.
Уменьшение введения плохо метаболизируемых веществ достигается с помощью модификаций диеты. Данный метод используется у больных с болезнями обмена аминокислот, нарушениями в цикле синтеза мочевины, при некоторых органических ацидуриях и митохондриальных энцефаломиопатиях. В лечении нарушений окисления жирных кислот применяется диетотерапия с ограничением жиров (до 15% калорийности) и обогащением углеводами (до 65% калорийности).
Связывание токсичного метаболита другими веществами и выведение его из организма в виде нетоксичных соединений реализуется посредством применения специальных лекарственных средств. Так, при гипераммониемии используются бензоат, фенилацетат и фенилбутират натрия. Бензоат натрия связывается с глицином и в виде нетоксичного соединения выводится из организма. При неэффективности данных препаратов используется гемо- или перитонеальный диализ. Параллельно при гипераммониемии проводится резкое ограничение в диете белка и внутривенно вводится глюкоза. Использование данных методик лечения привело к тому, что 93% детей с нарушениями цикла синтеза мочевины доживают до 1 года. При изовалериановой ацидемии с целью выведения токсичных метаболитов используется глицин, при дефектах -окисления жирных кислот — карнитин каксредство, повышающее интенсивность внутриклеточного транспорта жирных кислот.
К факторам, провоцирующим ухудшение течения некоторых дисметаболических заболеваний (например, дефицита ацил-СоА-дегидрогеназ) в неонатальном периоде и после него, относятся длительное голодание, избыток белков и жиров в диете и интеркуррентные заболевания. Нередко они каскад необратимых метаболических нарушений. Поэтому при многих наследственных болезнях обмена веществ следует избегать белковых или жировых пищевых нагрузок или голодания, проводить лечение интеркуррентных болезней.
Назначение высоких доз витаминов (тиамина 200-1000 мг/сут, рибофлавина 100 мг/cyт и никотинамида 1 г/сут) дает лечебный эффект при различных видах митохондриальных энцефаломиопатий. Некоторые рибофлавинзависимые формы множественного дефицита ацил-СоА-дегидрогеназ (глутаровая ацидемия, тип II) поддаются лечению высокими дозами рибофлавина (до 100 мг/сут).
Основные виды терапии наследственных болезней метаболизма с неонатальным дебютом представлены в табл. 7.
| Таблица 7. Особенности терапии при неонатальных формах наследственных заболеваний обмена веществ | |
| Заболевание | Лечение |
| Нарушения в цикле синтеза мочевины | Гемодиализ, перитонеальный диализВысококалорийная диета с резким ограничением содержания белка и добавками аминокислот Внутривенное или энтеральное введение бензоата натрия (250 мг/кг в сутки) или фенилацетата (250-500 мг/кг в сутки) или фенилбутирата (500 мг/кг в сутки) L-аргинин (400-800 мг/кг в сутки) Пересадка печени |
| Некетотическая гиперглицинемия | Внутривенное введение бензоата натрия (250 мг/кг в сутки)Пиридоксин (50 мг/кг в сутки) Противопоказан прием вальпроатов |
| Болезнь с запахом мочи кленового сиропа | Гемодиализ, перитонеальный диализВысококалорийная диета с ограничением аминокислот с разветвленной цепью (40-60 мг лейцина на 1 кг массы) Тиамин (50 мг — 500 мг/сут) |
| Изовалериановая ацидемия | Диета с ограничением содержания белкаL-карнитин (100 мг/сутки) Глицин (до 250 мг/кг/сут) |
| Метилмалоновая ацидурия | Диета с резким ограничением содержания белка (изолейцин, валин, метионин, треонин)Гидроксикобаламин, аденозилкобаламин (15 мг/сут) L-карнитин (100 мг/кг в сутки) Метронидазол (10-15 мг/сут) |
| Пропионовая ацидемия | Гемодиализ, перитонеальный диализДиета с резким ограничением содержания белка (изолейцин, валин, метионин, треонин) L-карнитин (100 мг/кг в сутки) Биотин (5-10 мг/сут) Метронидазол (10-15 мг/сут) |
| Дефицит 3-гидро-3-метилглутарил-СоА-лиазы | Частое кормление для исключения голоданияДиета с низким содержанием белка и липидов (углеводное питание) L-карнитин (100 мг/кг в сутки) |
| Дефицит фруктозо-1,6-бифосфатазы | Диета с исключением фруктозыВнутривенное введение глюкозы и бикарбоната |
| Дефицит пируватдегидрогеназного комплекса | Диета с высоким содержанием жиров и низким — углеводовL-карнитин (100 мг/кг в сутки) Тиамин (до 200 мг/сут) Дихлорацетат (35-50 мг/кг в сутки) Липоевая кислота (до 500 мг/сут) |
| Дефицит пируваткарбоксилазы | Диета с высоким содержанием жиров и низким — углеводовБиотин (10 мг/сут) Аспартатовая кислота |
| Дефицит I комплекса дыхательной цепи митохондрий | Диета с ограничением углеводовНикотинамид (до 500 мг/сут) Рибофлавин (до 100 мг/сут) L-карнитин (50-100 мг/кг в сутки) Дихлорацетат (35-50 мг/кг в сутки) |
| Множественная недостаточность ацил-СоА-дегидрогеназ жирных кислот | Диета с низким содержанием белка и липидовРибофлавин (100-300 мг/сут) L -карнитин (100 мг/кг в сутки) |
| Дефицит ацил-СоА-дегидрогеназ жирных кислот со средней длиной углеродной цепи | Частое кормление для исключения голоданияДиета со сниженным содержанием жиров и повышенным — углеводов L-карнитин (100 мг/кг в сутки) Рибофлавин (100-300 мг/сут) Внутривенное введение растворов глюкозы и бикарбоната натрия |
Значение своевременной диагностики врожденных дефектов метаболизма чрезвычайно велико. К доступным скрининговым методикам при наследственных болезнях обмена веществ с проявлениями в неонатальном периоде относятся определение содержания лактата, глюкозы и аммония в крови, органических кислот и аминокислот в моче. Несвоевременное установление диагноза и отсутствие адекватного лечения может привести к смерти новорожденного или к тяжелой неврологической инвалидности. Правильная трактовка патологии способна обеспечить оптимальное нервно-психическое развитие ребенка и оказать членам его семьи своевременную специализированную медико-генетическую помощь.
Литература
1. Вельтищев Ю.Е., Казанцева Л.З., Семячкина А.Н. Наследственные болезни обмена. Наследственная патология человека. Под общей редакцией Ю.Е. Вельтищева, Н.П. Бочкова. М 1992; 41- 101.
2. The Neurology of Neonatal Hereditary Metabolic Diseases. Neurology of Hereditary Metabolic Diseases of Children. Ed. G.Lyon, R.D. Adams, E.H. Kolodny. New York: McGraw-Hill 1996; 6-44.
3. Наследственные болезни и синдромы у детей с нарушениями нервно-психического развития. Под редакцией П.А. Темина, Л.З. Казанцевой. В печати.
4. Вельтищев Ю.Е., Темин П.А., Белоусова Е.Д. Митохондриальные болезни, обусловленные мутациями ядерной ДНК. Наследственные болезни нервной системы. М: Медицина 1998; 409- 469.
5. Николаева Е.А. Органические ацидемии, сопровождающиеся судорожным синдромом. Диагностика и лечение эпилепсии у детей. Можайск: Терра 1997; 395-426.
6. Metabolic Diseases of the Nervous System. Textbook of Child Neurology. Ed. J.H. Menkes. Baltimore: Williams and Wilkins 1995; 29-151.
7. Metabolic and Degenerative Diseases of the Central Nervous System. Pathology, Biochemistry, and Genetics. Ed. J. Cervos-Navarro, H. Urich. San Diego: Academic Press 1995; 873.
Авторы: Белоусова Е. Д. , Никанорова М. Ю., Николаева Е. А.
Источник: Российский вестник перинатологии и педиатрии, 2000, N6, с.12-19
Источник