Пищевая химия: учебник для студентов вузов
Лабораторная работа 39. Флуорометрический метод определения тиамина
В основе флуорометрического 5 метода анализа витамина В 1 в пищевых продуктах лежит реакция окисления в щелочной среде тиамина до тиохрома, который в УФ-свете имеет характерную синюю флуоресценцию, исчезающую при подкислении и вновь возникающую при подщелачивании. В качестве специфического окислителя применяют гексацианоферрат ( III ) калия.
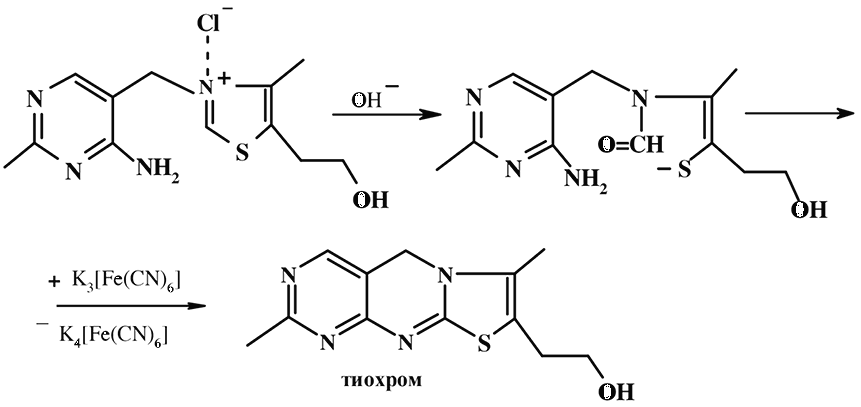
Образовавшийся желтого цвета тиохром экстрагируют растворителем, не смешивающимся с водой (изобутиловый, изоамиловый спирты). Флуоресценцию полученного раствора измеряют на флуорометре.
Анализ пищевых продуктов, представляющих собой сложные многокомпонентные смеси, требует предварительного разделения и дальнейшую очистку всех фракций, содержащих витамин В 1 . Поэтому первоначально проводят кислотный, а затем ферментативный гидролиз, в результате чего происходит высвобождение и перевод всех форм витамина В 1 в тиамин.
Полученный гидролизат подвергают хроматографическому разделению на катионите СДВ-3, используя в качестве элюента 25%-ный раствор хлорида калия, приготовленный на 0,1 н растворе соляной кислоты. При анализе несложных пищевых систем гидролизат промывают изобутиловым спиртом, который не растворяет витамин В 1 , но извлекает нежелательные флуоресцирующие примеси. Метод стандартизирован (ГОСТ–30627.5–98 «Продукты молочные для детского питания. Методы измерения массовой доли витамина В 1 »).
Ход анализа . Навеску анализируемого продукта (около 5–10 г), в зависимости от массовой доли витамина, тщательно растереть в фарфоровой ступке с 10–25 мл 0,1 н раствора соляной кислоты. Гомогенизированную массу количественно перенести в коническую колбу вместимостью 250 мл, используя тот же растворитель (общий объем раствора не должен превышать 150 мл). Поместить колбу в кипящую водяную баню.
По истечении 45 минут колбу вынуть из бани, охладить до температуры 35–40 о С и довести рН среды гидролизата до 4,5, используя насыщенный раствор уксуснокислого натрия (около 5–7 мл). Внести в колбу 0,1 г ферментного препарата амилоризина П10Х (пектаваморина П10Х), 0,5 мл толуола и выдержать полученную смесь в термостате при температуре 37 о С. Спустя 14–16 часов содержимое колбы охладить до комнатной температуры, количественно перенести в мерную колбу вместимостью 250 мл, довести объем смеси до метки дистиллированной водой и отфильтровать.
Для удаления флуоресцирующих примесей, мешающих анализу, отмерить в делительную воронку 25 мл фильтрата и добавить 25 мл изобутилового спирта. Воронку закрыть пробкой и поместить в вибросмеситель для экстракции на 5 минут. Дождаться полного разделения слоев жидкостей, верхний слой, содержащий флуоресцирующие примеси, оставить. Нижний (водный) слой собрать в мерную колбу вместимостью 25 мл и довести объем раствора до метки дистиллированной водой.
Окисление тиамина до тиохрома . В четыре делительные воронки вместимостью 50 мл отмерить по 5 мл очищенного от флуоресцирующих примесей фильтрата. Добавить в каждую воронку по 2 мл этилового спирта и перемешать. В первую и вторую делительные воронки внести по 1,2 мл окислительной смеси, состоящей из раствора гексацианоферрата ( III ) калия и гидрокисида калия. В третью и четвертую воронки прилить по 1,2 мл 30%-го раствора гидроксида натрия (холостые пробы). Закрыть воронки пробками, поместить на вибросмеситель и экстрагировать в течение 2 минут.
Во все воронки добавить по 13 мл изобутилового спирта и продолжить встряхивание в течение 3 минут. Оставить воронки в темном месте на 10 минут для разделения слоев жидкости. Нижний слой (водный) слить, а к верхнему слою (изобутилового спирта) добавить по 2 мл этилового спирта для осветления. Смесь встряхнуть, дать отстояться и отфильтровать через бумажный фильтр в кювету флуориметра.
Измерение флуоресценции стандартного раствора тиамина . Отмерить в четыре делительные воронки (вместимостью 50 мл) по 5 мл рабочего раствора тиамин хлорида (1 мкг/мл), добавить по 2 мл этилового спирта и перемешать. Провести окисление стандартных растворов тиамина также как и анализируемых растворов. Измерить флуоресценцию стандартных растворов тиамина после окисления и соответствующих холостых проб (без окислителя), а затем флуоресценцию анализируемых растворов и соответствующих им холостых проб на флуорометре со светофильтрами, имеющими максимум пропускания в области 320–390 нм (В 1 -первичный, возбуждающего света) и 400–580 нм (В 1 -вторичный, излучаемого света). Массовую долю витамина В 1 ( С В1 , млн –1 ) рассчитать по формуле
C B 1 = ( A — A 1 ) × m × V × V 1 ( A C — A C 1 ) × m 1 × V 2 × V 3 ,
где А – среднеарифметическое значение показаний флуорометра для анализируемого продукта ;
А 1 – среднеарифметическое значение показаний флуорометра для холостой пробы (без окислителя) анализируемого продукта ;
А С – среднеарифметическое значение показаний флуорометра для стандартного раствора тиамина;
A C 1 – среднеарифметическое значение показаний флуорометра для холостой пробы (без окислителя) стандартного раствора тиамина;
m – масса тиамина в 1 мл рабочего раствора, мкг;
m 1 – масса анализируемого продукта, г ;
V – общий объем гидролизата, 250 мл;
V 1 – объем гидролизата, взятого для очистки от примесей, 25 мл;
V 2 – объем анализируемого раствора, взятого на окисление тиамина в тиохром, 5 мл;
V 3 – объем фильтрата после стадии очистки, 25 мл.
Необходимые реактивы, посуда, оборудование :
§ 0,1 н раствор соляной кислоты, 30%-ный раствор гидроксида калия, 1%-ный раствор гексацианоферрата ( III ) калия (красной кровяной соли), окислительная смесь (гексацианоферрата ( III ) калия в гидроксиде калия), основной раствор тиамин хлорида (0,1 мг/мл), рабочий раствор тиамин хлорида (1 мкг/мл), насыщенный раствор уксуснокислого натрия, изобутиловый спирт, этиловый спирт, толуол, ферментный препарат амилоризин П10Х или пектаваморин П10Х (сухой препарат, полученный переосаждением);
§ пипетки, делительные воронки, фарфоровые ступки с пестиками, стеклянные воронки, мерные колбы, колбы плоскодонные;
§ аналитические весы, флуорометр (с набором светофильтров), рН- метр, водяная баня, термостат, вибросмеситель.
1%-ный раствор гексацианоферрата ( III ) калия ( K 3 [ Fe ( CN ) 6 ] ) . Точную навеску (1 г) гексацианоферрата ( III ) калия растворить в 30 мл дистиллированной воды в мерной колбе вместимостью 100 мл и довести объем раствора до метки. Раствор перемешать и перелить в склянку из темного стекла (хранить не более двух суток). Концентрация раствора – 0,01 г/мл.
Окислительная смесь . В стаканчик отмерить пипеткой 2 мл 1%-го раствора гексацианоферрата ( III ) калия, добавить 10 мл 30%-го раствора гидроксида калия и перемешать. Срок хранения окислительной смеси не более 3 ч.
Насыщенный раствор уксуснокислого натрия . В мерной колбе вместимостью 500 мл взвесить 200 г уксуснокислого натрия ( C 2 H 3 O 2 Na × 3 H 2 O ), растворить соль в 200 мл дистиллированной воды и довести объем до метки тем же растворителем.
Очистка изобутилового спирта . К 1 л изобутилового спирта добавить 15–20 г активированного угля, встряхнуть и продолжить встряхивать в течение 30 минут. Раствор оставить на сутки, затем отфильтровать через бумажный фильтр и перегнать.
Основной раствор тиамин хлорида ( 0,1 мг/мл ) . Точную навеску тиамин хлорида (0,1 г), высушенного в эксикаторе над серной кислотой в течение 10 суток, растворить в мерной колбе вместимостью 1 л в 200 мл дистиллированной воде и довести объем раствора до метки. Раствор хранить в склянке из темного стекла в холодильнике не более 2 месяцев.
Рабочий раствор тиамин хлорида ( 1 мкг/мл ) . В мерную колбу вместимостью 100 мл пипеткой отмерить 1 мл основного раствора тиамина (0,1 мг/мл) и довести объем до метки. Хранить не более суток.
5 Флуоресценция (разновидность люминесценции) представляет собой вторичное излучение молекул в короткий период времени непосредственно после поглощения инициирующего ультрафиолетового излучения. Длина волны второго излучения больше, чем инициирующего. Для разбавленных растворов наблюдается линейная зависимость интенсивности флуоресценции от концентрации.
Источник
Работа 78. Определение содержания тиамина и рибофлавина флуориметрическим методом в поливитаминных препаратах
Реактивы. Соляная кислота, 0,1 М раствор; окислительная смесь * ; Н-бутанол; этанол, 96%-ный; тиамин, стандартный раствор концентрации 10 мкг/мл; уксусная кислота, ледяная; перманганат калия, 4%-ный раствор; гидроксид водорода, 3%-ный раствор; гидросульфит натрия, порошок; рибофлавин, стандартный раствор концентрации 0,005 мг/мл.
Оборудование. Штатив с пробирками; пенициллиновые флакончики с полиэтиленовыми пробками; пипетки вместимостью 1 и 5 мл; мерный цилиндр вместимостью 50 мл; ступка с пестиком; флуориметр ЭФ-3 или БИАН.
Материал. Драже поливитаминов.
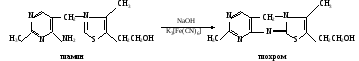
а. Определение содержания тиамина. Метод основан на способности тиамина окисляться гексацианоферратом (III) калия в щелочной среде в тиохром, который после извлечения его из раствора бутиловым спиртом дает в ультрафиолетовом свете сине-голубую флуоресценцию:
Ход определения. Драже поливитаминов разминают в ступке, добавляя 30 мл раствора соляной кислоты, и тщательно перемешивают.
В один флакончик (контроль) вносят 5 мл соляной кислоты, во второй (опыт) – 1 мл водного экстракта драже витаминов и 4 мл дистиллированной воды, в третью (стандарт) – 5 мл раствора тиамина.
Во все флакончики приливают по 1,5 мл окислительной смеси и осторожно встряхивают их до полного перемешивания. Затем добавляют в них по 5 мл бутанола, плотно закрывают пробками и интенсивно встряхивают 5 мин. После расслоения жидкости осторожно прибавляют по 0,5 мл этанола (для просветления бутанола).
Осторожно сливают просветленный бутанольный слой в кювету флуориметра и измеряют интенсивность флуоресценции опытной и контрольной проб со стандартным раствором тиамина.
Расчет проводят по формуле
где х – содержание тиамина в драже, мг;
Еоп – показания флуориметра для опытной пробы;
Ек – показания флуориметра для контрольной пробы;
Ест – показания флуориметра для стандартной пробы;
0,01 – концентрация тиамина в стандартном растворе, мг/мл;
30 – объем экстракта драже, мл;
1 – объем экстракта, взятого на исследование, мл;
5,5 – объем пробы, просветленной этанолом, мл.
б. Определение содержания рибофлавина. Принцип метода см. работу 73, е.
Ход определения. Драже поливитаминов разминают в ступке, добавляя 30 мл раствора соляной кислоты, и тщательно перемешивают.
В одну пробирку вносят 7 мл дистиллированной воды, во вторую (опытную) – 2 мл экстракта драже и 5 мл дистиллированной воды, в третью (стандартная) – 1 мл стандартного раствора рибофлавина и 6 мл воды.
Во все пробирки приливают по 10 капель ледяной уксусной кислоты и по 1,5 мл раствора перманганата калия (для окисления посторонних флуоресцирующих веществ).
Содержимое пробирок встряхивают и добавляют по каплям (примерно 5 капель) гидроксид водорода при постоянном помешивании стеклянной палочкой до полного просветления жидкости. Растворы отстаивают 5 мин, до прекращения выделения пузырьков газа. Сливают жидкость в кюветы флуориметра и измеряют интенсивность флуоресценции всех проб.
Расчет проводят по формуле
где х – содержание рибофлавина в драже, мг;
Еоп – показания флуориметра для опытной пробы;
Ек – показания флуориметра для контрольной пробы;
Ест – показания флуориметра для стандартной пробы;
30 – объем экстракта драже, мл;
2 – объем экстракта драже, взятого на исследование, мл;
0,005 – концентрация рибофлавина в стандартном растворе, мг/мл;
7 – объем флуориметрируемых проб, мл.
Оформление работы. Рассчитать содержание исследуемых витаминов в драже и сделать вывод о возможности практического использования флуориметрического метода.
Практическое значение работы. Флуориметрические методы определения тиамина и рибофлавина применяются для определения этих витаминов в пищевых продуктах, лекарственных растениях и готовых лекарственных препаратах, а также для изучения обеспеченности ими организма. Обеспеченность этими витаминами может быть определена по их уровню в крови и по экскреции с мочой. Низкое содержание витаминов в организме наблюдается при гиповитаминозах, болезнях печени, сердечно-сосудистых заболеваниях, заболеваниях желудочно-кишечного тракта и других патологических состояниях.
Источник
Флуориметрический метод анализа
Нелинейная флуориметрия сложных органических веществ. Приборы для проведения флуориметрического метода в фармацевтическом анализе. Общая фармакопейная статья Российской Федерации. Флуориметрическое определение кверцетина в лекарственных формах рутина.
| Рубрика | Химия |
| Вид | курсовая работа |
| Язык | русский |
| Дата добавления | 07.06.2018 |
| Размер файла | 139,0 K |

Отправить свою хорошую работу в базу знаний просто. Используйте форму, расположенную ниже
Студенты, аспиранты, молодые ученые, использующие базу знаний в своей учебе и работе, будут вам очень благодарны.
Размещено на http://www.allbest.ru/
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РЕСПУБЛИКИ УЗБЕКИСТАН
ТАШКЕНТСКИЙ ФАРМАЦЕВТИЧЕСКИЙ ИНСТИТУТ
КАФЕДРА ФАРМАЦЕВТИЧЕСКОЙ ХИМИИ
По предмету: Фармацевтическая химия
По теме: Флуориметрический метод анализа
Руководитель: доцент Юнусходжаева Н.А.
Подготовил: студент III-курса
Промышленной фармации 6/2 -группа
1. Теоретические основы флуориметрического анализа
1.1 Понятие и сущность флуориметрии
1.2 Физическая сторона флуориметрического анализа
1.3 Нелинейная флуориметрия сложных органических веществ
1.4 Обратные задачи флуориметрии насыщения
1.5 Приборы для проведения флуориметрического метода в фармацевтическом анализе
1.6 Общая фармакопейная статья Российской Федерации
2. Экспериментальная часть
2.1 Определение содержания рибофлавина (витамина В2) методом флуориметрии
2.2 Статистический расчёт содержания рибофлавина (витамина В2) методом флуориметрии
2.3 Флуориметрическое определение содержания родамина
2.4. Флуориметрическое определение кверцетина в лекарственных формах рутина
2.5. Флуориметрическое определение 2-амино-4-окси-6- птеридинкарбоновой кислоты в лекарственных формах фолиевой кислоты
Список использованных литератур
В современном мире проблема качества лекарственных средств является важнейшим фактором повышения уровня жизни, экономической, социальной и экологической безопасности.
Критерии качества лекарственных средств, установленные Всемирной организацией здравоохранения включают в числе обязательных элементов соответствие требованиям спецификаций качества, устанавливающим тщательно отобранные нормы, методы испытаний и др. Как правило, спецификации качества содержатся в таких нормативных документах, как общие фармакопейные статьи (ОФС), фармакопейные статьи (ФС), фармакопейные статьи предприятия (ФСП), а также ГОСТы, ОСТы, ТУ и др.
Это предусматривает, гармонизацию требований, предъявляемых различными производителями одних и тех же ЛС к их качеству и методикам определения этого качества.
Анализ нормативных документов зарубежных стран и отечественных производителей свидетельствует о том, что во всем мире прослеживается четкая тенденция к внедрению в практику фармацевтического анализа современных инструментальных методов, обеспечивающих специфичность аналитических методик. Это и обуславливает тот факт, что в последние годы при оценке качества ЛС широкое распространение получили различные методы физико-химического анализа, такие как спектрофотометрия, хроматография, ЯМР-спектроскопия, флуориметрия, капиллярный электрофорез и другие.
До сих пор в большинстве случаев для количественного определения большого количества препаратов применяются традиционные титриметрические методы. Несмотря на неоспоримые достоинства титриметрии, этот метод имеет ряд ограничений, к числу которых следует отнести: невозможность получения достоверного результата в присутствии других компонентов, входящих в состав лекарственного средства; высокая лабильность и способность к окислению.
В то же время, такой метод анализа как флуоресцентный метод характеризуется высокой специфичностью, позволяющей осуществлять оценку качества определяемого вещества в присутствии других компонентов и чувствительностью, близкой, в некоторых случаях, к чувствительности радиохимических методов. Все это дает возможность для использования метода флуориметрии при проведении качественного и количественного анализа не только субстанций и монопрепаратов, но и комплексных витаминосодержащих лекарственных средств, лекарственного растительного сырья, растительных лекарственных средств и препаратов.
Цель работы — характеристика методик флуоресцентного качественного и количественного анализа лекарственных веществ.
— рассмотреть основные теоретические положения описываемого физико-химического метода анализа,
— проанализировать применение изучаемого метода в анализе лекарственных средств определение подлинности, чистоты и допустимых примесей, количественного содержания,
— выявить химические свойства лекарственных средств, обусловливающие возможность использования изучаемого метода.
1. Теоретические основы флуориметрического анализа
Преимущества, которые дает флуориметрия в анализе лекарственных средств в различных объектах, способствовали введению этого метода в перечень фармакопейных.
В функциональном анализе фармацевтических соединений предполагается обычно, что молекулу органического соединения можно рассматривать как сумму практически независимых функциональных групп и, следовательно, принимается, что физические и химические свойства соединения определяются свойствами этих функциональных групп. При проведении идентификации сложных молекул, несомненно, следует учитывать взаимное влияние функциональных групп, которое может вызвать неожиданное изменение свойств этих групп, а также отклонения наблюдаемых свойств от теоретически ожидаемых.
Методы флуоресцентного анализа имеют важное самостоятельное значение особенно в промышленности при получении фармацевтических препаратов, так как проведение функционального анализа — важная и часто необходимая стадия при определении качественного состава сложных смесей. органический флуориметрический кверцетин рутин
Учитывая вышесказанное, показан поиск теоретически обоснованного подхода к разработке реакций флуоресцентного определения. В основу подхода была положена идея получения конкретного производного, флуоресцирующего в видимой области спектра. Результатом исследования стали флуоресцентные реакции и разработанные на их основе методики обнаружения и количественного определения примесей в лекарственных препаратах.
Флуоресценция — излучательный переход из возбужденного состояния с синглетного уровня S1 в основное состояние S0: S1 > S0 + hнфл. Такие переходы квантовомеханически «разрешены», а типичная величина скоростей испускания для них
108 с-1. Высокие значения скоростей испускания приводят к временам затухания
10-11 — 10-8 сек. Время жизни — это средний период времени, в течение которого флуорофор находится в возбужденном состоянии. Для явления флуоресценции известно несколько основных характеристик, такие как стоксов сдвиг, квантовый выход, энергетический выход, время затухания. Стоксовым сдвигом называется сдвиг испускания относительно поглощения в сторону больших длин волн, то есть наблюдается потеря энергии (это явление впервые наблюдал Стокс). Вследствие того, что практически всегда при поглощении и испускании света осуществляется один и тот же электронный переход, соблюдение правила Стокса требует выполнение следующего соотношения hнвозб — h нлюм > 0. Впоследствии Ломмель предложил такую формулировку, которая используется в современной науке: спектр излучения в целом и его максимум всегда сдвинуты по сравнению со спектром поглощения и его максимумом в сторону длинных волн. Эта зависимость получила название закона Стокса — Ломмеля, который может быть записан в следующем виде: hнmax люм -9 10 -8 с. Короткое время жизни флуоресценции отличает этот тип люминесценции от фосфоресценции, которая представляет собой долгоживущее свечение, имеющее время жизни от 10 -3 с до нескольких мин.
Метод флуориметрии в 10 100 раз чувствительнее абсорбционной спектрофотометрии, но флуоресцентными свойствами обладает только ограниченный круг соединений: ароматические, особенно с конденсированными структурами, гетероциклические и карбонильные соединения. Из фармацевтических субстанций определению методом флуориметрии подлежат аминокислоты (фенилаланин, триптофан, тирозин), алкалоиды (стрихнин, резерпин, хинин), витамины (фолиевая кислота, рибофлавин, ретинола ацетат), стероидные гормоны (этинилэстрадиол).
Интенсивность флуоресценции измеряется в условных единицах, пропорциональных отклику детектора и обозначается символом I.
Спектр испускания флуоресценции представляет собой зависимость интенсивности флуоресценции от длины волны (в нм) или частоты (в см -1 ) при заданной длине волны возбуждения. Спектр возбуждения флуоресценции представляет собой зависимость интенсивности излучения в максимуме испускания флуорофора от длины волны или частоты возбуждающего света. При этом спектр возбуждения обычно совпадает со спектром поглощения, так же как и интенсивность флуоресценции пропорциональна светопоглощению. Комбинирование спектров испускания, полученных при различных длинах волн возбуждения, даёт трёхмерную карту испускания.
1.2 Физическая сторона флуориметрического анализа
Поглощение света, безусловно, является необходимым условием флуоресценции, но в тех случаях, когда поглощение раствора слишком высоко, световой поток не проходит через него и не может служить источником возбуждения. Как известно, при высоких концентрациях участки раствора, расположенные ближе к источнику возбуждения, поглощают большее количество света, чем дальше расположенные. Интенсивность освещения раствора и, соответственно, излучение прогрессивно убывают по мере удаления от источника (внутреннее экранирование).
Процесс флуоресценции веществ подчиняется законам светопоглощения, как и все оптические методы анализа: интенсивность проходящего через раствор вещества светового потока уменьшается согласно закону Бера — Ламберта:
где: I — интенсивность светового потока, проходящего через раствор;
Iо- интенсивность падающего света;
К — коэффициент поглощения раствора;
С — концентрация вещества;
L — длина пути светового потока в поглощающем растворе вещества.
Интенсивность флуоресценции (испускаемой во всех направлениях) зависит от количества поглощаемого флуоресцирующим объектом света и квантового выхода флуоресценции :
где: F — общая интенсивность флуоресценции;
D — оптическая плотность раствора.
При малых величинах D« 0,1 формула имеет следующий вид:
F = 10 ф(1 — (1 — D + D2/2′ — D3/3′ In), или F = I0*D
Таким образом, зависимость F от D (с учетом значения квантового выхода и интенсивности возбуждающего света) является линейной, что более удобно для экспериментальных исследований. Переход к линейной зависимости позволяет рассчитывать концентрацию флуорофоров по законам светопоглощения.
Однако необходимо вычислить минимальное значения оптической плотности раствора D0, при которых ошибка измерения интенсивности флуоресценции не превышает некоторой заданной величины.
Как правило, линейная зависимость флуоресценции от концентрации раствора наблюдается до тех пор, пока количество флуоресцирующего вещества не становится настолько большим, что раствор начинает поглощать значительное количество возбуждающего света. Боуен и Уокс показали, что для получения линейной зависимости раствор должен поглощать менее 5% возбуждающего света. Поэтому Dn = 2,0×0,05 = 0,1, то есть предел линейной зависимости флуоресценции от концентрации определяется достижением оптической плотности D = 0,1.
Поскольку D = KCL, то одной и той же оптической плотности могут соответствовать различные концентрации в зависимости от толщины поглощающего слоя. Следовательно, использование кювет с различной толщиной рабочего слоя позволяет применять флуориметрию для измерения высоких концентраций без предварительного разбавления проб. При практическом использовании этого приема выявлена закономерность, связывающая изменения толщины рабочего слоя кюветы и предела обнаружения методики: при уменьшении рабочего слоя в 2 раза верхний пределе линейности калибровочного графика увеличивается в среднем в 1,4 раза.
Измерения отношения интенсивности флуоресценции вещества к интенсивности облучающего пробу светового потока (F/I0) позволяет определять величину, подобную оптической плотности в спектрофотометрии. Эта величина зависит только от концентрации исследуемых веществ при использовании стандартных кювет, что позволяет стандартизовать измерения флуоресценции и избавится от необходимости использования стандартных растворов.
1.3 Нелинейная флуориметрия сложных органических веществ
Сложные органические соединения играют большую роль в природе, широко используются в самых различных технологиях. Поэтому разработка методов и средств их обнаружения, измерения концентрации и определения состояния (то есть методов диагностики) была и остается актуальной научной и технической проблемой. Особый интерес представляют сложные органические соединения, которые входят в состав природных сред и живых организмов или которые попадают в эти объекты, а иногда специально вводятся в них с теми или иными целями.
Очевидно, что наиболее ценными являются такие методы и средства, которые обеспечивают диагностику без разрушения объекта (то есть in vivo), непосредственно в среде обитания сложного органического соединения (то есть in situ), с высокой чувствительностью, экспрессно, а в ряде случаев (например, в системах экологического мониторинга) дистанционно.
Природа преподнесла нам подарок: многие органические соединения обладают способностью флуоресцировать при оптическом возбуждении с квантовым выходом, достаточным для решения диагностических задач. Все они обладают полосами флуоресценции, положение, форма и интенсивность которых могут быть использованы для диагностики указанных соединений.
Флуориметрия как метод спектрального анализа и диагностики сложных органических соединений обладает достоинствами, делающими ее чрезвычайно привлекательной. Во-первых, полоса флуоресценции смещена (и иногда весьма значительно) в длинноволновую область спектра относительно полосы поглощения, что позволяет при регистрации флуоресценции отстраиваться от фона, создаваемого упругим рассеянием фотонов на неоднородностях среды (рассеянием Рэлея и Ми), и эффективно подавлять этот фон. Во-вторых, сечение флуоресценции для хорошо флуоресцирующих соединений настолько велико, что позволяет регистрировать флуоресценцию сред с чрезвычайно низкой концентрацией флуоресцирующих молекул.
Замена обычных источников возбуждающего излучения на лазеры значительно улучшает характеристики флуоресцентного анализа в классическом варианте (в частности, еще больше поднимает чувствительность). Но главная, принципиально новая возможность, которая открывается с применением лазеров во флуориметрии, связана с насыщением флуоресценции — нелинейной зависимостью числа фотонов флуоресценции от плотности потока фотонов возбуждающего излучения. На этом основана нелинейная флуориметрия. Но прежде чем перейти к изложению физических основ этого нового метода спектроскопии, посмотрим, так ли велика необходимость в этом методе. А для этого укажем на трудности (и подчас весьма серьезные), с которыми сталкивается классическая (линейная) флуориметрия. Для разных объектов в различных задачах диагностики эти трудности проявляются по-разному. Для определенности рассмотрим, какие препятствия возникают на пути решения задач диагностики природных органических комплексов (ПОК) в водных средах. Первое препятствие — большая ширина полос флуоресценции ПОК и перекрытие полос разных ПОК, одновременно присутствующих в природных водах. Определяющий вклад в спектр флуоресценции этих сред дают четыре класса ПОК: фотосинтезирующие организмы — фитопланктон, водное гумусовое вещество, аминокислоты в свободном состоянии или в составе белковых соединений, нефти в виде пленки на поверхности, эмульсии и в растворенной форме в объеме воды.
Второе препятствие — идентичность или близость полос флуоресценции представителей одного класса ПОК.
Третье препятствие. Изменение состояния ПОК, как правило, практически не отражается на форме и положении полос флуоресценции, но влияет на ее интенсивность. Это затрудняет определение концентрации ПОК по интенсивности флуоресценции и не дает возможности проводить диагностику состояния ПОК (например, фотосинтетической активности водорослей).
Преодолеть указанные препятствия и решить в полной мере проблему диагностики ПОК невозможно в рамках только феноменологического подхода, то есть оперируя лишь спектрами излучения и возбуждения флуоресценции, хотя, конечно, эти спектры дают большой объем информации. Необходимо выходить на молекулярный уровень и в дополнение к спектрам использовать молекулярные фотофизические параметры: сечения поглощения, флуоресценции, возбуждения флуоресцирующих молекул, константы скоростей внутримолекулярных переходов и межмолекулярного переноса энергии. При этом указанные параметры необходимо измерять in vivo и in situ, причем в условиях дефицита информации о локальных концентрациях молекул в ПОК.
В настоящее время единственный способ измерения указанных параметров — нелинейная флуориметрия (флуориметрия насыщения). Важно подчеркнуть, что на этом пути решаются не только прикладные задачи диагностики ПОК, но и фундаментальные проблемы: установление механизмов
Причин для этого может буть несколько более физически понятная состоит в следующем. Если плотность потока фотонов F столь велика, что среднее время между их попаданиями в площадку, численно равную сечению поглощения молекулы oabs, меньше времени жизни молекулы в возбужденном состоянии, то молекула не успевает вернуться в основное состояние к моменту попадания в нее следующего фотона и этот фотон пролетает мимо молекулы — мы (при величине квантового выхода флуоресценции л = 1) лишаемся одного фотона флуоресценции (если слой среды оптически тонкий, то есть oabs InO Т1.
Параметр kj (его размерность с»1) характеризует вероятность перехода с уровня на уровень j таким образом, что если п — концентрация молекул в исходном состоянии то kipi — число молекул (в единице объема), перешедших за 1 секунду из состояния в состояние j. В рассматриваемой модели имеются три канала дезактивации уровня S1, которые характеризуются параметрами к31, к31 и к32. Поэтому полное число молекул (в единице объема), покидающих уровень S1 в 1 секунду, равно кЗпЗ и, следовательно, хЗ = к-1 — время жизни этого уровня.
Число фотонов флуоресценции, испущенных элементом объема dV= dx*dy*dz в интервале времени от t до t + dt. [19]
1.4 Обратные задачи флуориметрии насыщения
Зная характеристики кривой насыщения, можно в принципе определить фотофизические параметры молекулы или комплекса. Задачи такого рода называются обратными. В общем случае они являются некорректными.
Однако, используя априорную информацию о фотофизических процессах, формирующих флуоресценцию сложных органических соединений и комплексов, можно ограничить число параметров модели и пределы их изменения так, чтобы задача стала корректной по А.Н. Тихонову, то есть удовлетворяла трем условиям: решение существует, решение единственно и устойчиво. В нашей задаче устойчивость гарантирована ограниченностью числа восстанавливаемых параметров: в рассмотренных к настоящему времени обратных задачах нелинейной флуориметрии число определяемых параметров варьировали от одного до трех и в будущем оно едва ли превысит пять. [3]
Что касается единственности решения, то она доказана для трехпараметрической модели, описываемой системой кинетических уравнений с определяемыми параметрами. Таким образом, эта обратная задача является корректной по А.Н. Тихонову. Есть все основания полагать, что добавление четвертого параметра — константы скорости синглет-синглетной аннигиляции g не приведет к утрате этого свойства, хотя здесь требуется отдельное доказательство. [24]
Теоретически устойчивая обратная задача может стать неустойчивой на практике.
Причинами практической неустойчивости являются погрешности (шумы) входных данных и отклонение реальных процессов от модели, по которой рассчитывают теоретические зависимости (в нашем случае кривые насыщения). Практическая неустойчивость возникает, когда шумы входных данных или отклонение процессов от модели превышают некоторые допустимые величины. Пороги возникновения практической неустойчивости зависят как от характера фотофизических процессов в объекте, так и от свойств алгоритма решения обратной задачи. В общем случае кривые насыщения флуоресценции — исключительно неблагоприятный класс входных данных для обратных задач. Это монотонные кривые без каких-либо резких элементов, за которые можно было бы «зацепиться». Для одного типа объектов кривые насыщения по форме мало отличаются друг от друга. [11]
Это приводит к тому, что практическая неустойчивость возникает уже при весьма низком уровне погрешностей входных данных: для двухпараметрической задачи это единицы процентов, для трехпараметрической менее процента, что трудно обеспечить в эксперименте. Однако это имеет место при использовании наиболее распространенного метода решения обратных задач — метода наименьших квадратов (МНК). В его основе лежит процедура минимизации функционала невязки между экспериментальными и теоретическими данными путем вариации искомых параметров в ограниченном объеме их возможных значений. Если экспериментальная кривая неидеальна (снята с большими погрешностями или не соответствует теоретической модели), одного интегрального параметра (невязки) оказывается недостаточно, чтобы найти наилучшую аппроксимирующую кривую, рассчитанную для значений параметров модели в допустимой (из физических соображений) области их значений. [20]
Некоторое время ситуация казалась тупиковой, и метод нелинейной флуориметрии мог умереть, не успев родиться. На помощь пришла современная компьютерная техника искусственных нейронных сетей (ИНС).
Уникальным свойством нейросетей является их способность обучаться на примерах и не просто запоминать, а обобщать представленную информацию, выявлять скрытые закономерности и классифицировать предъявляемые данные. В ИНС для операции с экспериментальными данными создается сеть нейронов, каждый из которых имеет некоторое количество входов и один выход. Математически нейрон можно описать как взвешенный сумматор:
где у — значение выходной величины, Т — передаточная функция (обычно нелинейная), xi — значения входных величин, w: — веса связей. ИНС состоит из нескольких слоев нейронов. Входной слой имеет количество нейронов, равное количеству элементов в наборе входных данных, в нашем случае количеству точек на кривой насыщения. Выходной слой должен иметь число нейронов, совпадающее с числом определяемых параметров. На первом этапе работы сети происходит ее обучение на известных данных с целью получения наилучшей матрицы весов w. [16]
Процесс обучения часто занимает продолжительное время, зато хорошо обученная сеть быстро и с высокой точностью выдает значения искомых параметров при предъявлении ей набора экспериментальных данных.
Применительно к нашей обратной задаче принципиальное отличие техники ИНС от традиционного метода наименьших квадратов состоит в том, что ИНС узнает кривую насыщения не по одному интегральному параметру (невязке), а по большому числу признаков. Набор этих признаков формируется в процессе тренировки сети и является ее секретом. Как показали модельные расчеты (численные компьютерные эксперименты), алгоритмы решения обратных задач, основанные на технике ИНС, уже сейчас обеспечивают вполне приемлемую практическую устойчивость решения, а ведь возможности ИНС в этой области далеко не исчерпаны. [25, 32]
1.5 Приборы для проведения флуориметрического метода в фармацевтическом анализе
Приборы для проведения флуориметрического метода в фармацевтическом анализе
Приборы, предназначенные для измерения флуоресценции, можно разделить на три категории: — флуориметры, флуоресцентные приставки к спектрофотометрам и спектрофлуориметры.
Каждый такой прибор включает в себя шесть основных узлов:
— источник возбуждающего света;
— селектор частоты возбуждающего света и частоты люминесценции;
— кюветное отделение, предназначенное для размещения кюветы с измеряемым образцом (стандартным образцом или пробой);
— фотоприемник люминесценции (фотоэлемент, фотоумножитель, фотодиод);
Во флуориметрах селекция частот возбуждающего света и света флуоресценции осуществляется с помощью светофильтров. Флуориметры применяют для проведения серийных анализов, где снижение селективности не является большим недостатком, а высокая чувствительность, напротив, представляет собой важное достоинство. [22]
При измерении флуоресценции с помощью флуориметров существенное значение имеет правильный выбор светофильтров, возбуждающего флуоресценцию (первичный светофильтр), и света флуоресценции (вторичный светофильтр). Первичный светофильтр должен пропускать свет в области поглощения определяемого вещества и не должен пропускать свет в области, отвечающей флуоресценции вещества. Вторичный светофильтр должен пропускать флуоресценцию, но возбуждающий свет должен им полностью поглощаться. Источниками возбуждающего света во флуориметрах обычно служат ртутные лампы низкого давления. С целью получения практически сплошного спектра излучения внутренние стенки этих ламп часто покрывают люминофором. [12, 34]
1.6 Общая фармакопейная статья российской федерации
Приборы. Для проведения флуориметрического анализа используют приборы двух типов: фильтрационный флуориметр и спектрофлуориметр.
Фильтрационный флуориметр состоит из источника излучения, первичного фильтра длин волн, камеры для образца, вторичного фильтра длин волн и системы детектирования флуоресценции. Как правило, детектор помещен под углом 90 о к возбуждающему световому потоку. Геометрия прямого угла предусматривает детектирование только произведенного флуоресцентного сигнала. Однако детектор все-таки получает часть возбуждающего излучения в результате рассеивающих свойств самого раствора, а также из-за присутствия в растворе твердых частиц. Для устранения этого остаточного рассеяния используются спектральные фильтры. Первичный фильтр отбирает коротковолновое излучение, способное к возбуждению испытуемых образцов, вторичный фильтр пропускает флуоресценцию в длинноволновой области, но блокирует рассеянное возбуждение.
Детекторы флуориметров преобразуют оптический сигнал в электрический с помощью фотоумножителей разных типов. Каждый тип детектора имеет специальные характеристики: спектральная область максимальной чувствительности, степень усиления, соотношение сигнал/шум.
Спектрофлуориметры отличаются от фильтрационных флуориметров тем, что вместо спектральных фильтров в них используются монохроматоры типа призмы или решетки. Эти приборы более предпочтительны для аналитических целей. В спектрофлуориметрах монохроматоры снабжены щелями. Чем уже щель, тем выше разрешение и спектральная чистота, но меньше чувствительность. Выбор размера щели определяется разделением между длинами волн возбуждающего и испускаемого излучения и необходимым уровнем чувствительности.
В качестве источников возбуждающего излучения в флуориметрах используют:
— ртутные лампы низкого давления, предоставляющие большое количество длин волн возбуждения, но не являющиеся источником излучения равномерного спектра;
— ксеноновые газоразрядные лампы, обеспечивающие высокоинтенсивное почти равномерное излучение в широком диапазоне спектра (300 — 800 нм) и достаточно интенсивное в коротковолновой области вплоть до 200 нм;
— лазеры, излучающие свет высокой интенсивности в очень узком интервале длин волн (не более 0,01 нм) и позволяющие благодаря этому не использовать монохроматоры или первичные светофильтры;
— светодиоды и светодиодные матрицы, излучающие свет в определённых диапазонах длин волн.
Для размещения анализируемых проб в флуориметрах используют, как правило, прямоугольные кварцевые кюветы, отполированные со всех 4 вертикальных сторон, иногда — цилиндрические кюветы или пробирки. Обычно объем испытуемых образцов составляет 2 — 3 мл, но к некоторым приборам прилагаются кюветы вместимостью от 100 до 300 мкл или капиллярные держатели для еще меньшего объема.
Измерение флуоресценции. Флуоресценцию определяют в растворах с концентрацией от 10 -5 М и менее, в диапазоне, для которого наблюдается прямая зависимость интенсивности флуоресценции от концентрации. При более высоких концентрациях всё более значительная часть поступающего света абсорбируется образцом вблизи поверхности кюветы, и линейная зависимость величины сигнала от концентрации определяемого вещества нарушается.
Все замеры интенсивности флуоресценции должны быть скорректированы с растворителем.
Интенсивность флуоресценции зависит от:
— величины рН испытуемого раствора,
— присутствия в растворе посторонних частиц,
— концентрации кислорода в испытуемом растворе,
Эффективность флуоресценции обратно пропорциональна температуре. Для некоторых веществ эффективность флуоресценции может снижаться на 1 2 % при повышении температуры на 1 о С. В таких случаях требуется термостатирование образцов.
Интенсивность и спектральное распределение флуоресценции зависит от растворителя. Многие соединения, флуоресцирующие в органических растворителях, фактически не флуоресцируют в воде.
Перед измерением флуоресценции из испытуемого раствора фильтрованием или центрифугированием должны быть удалены твёрдые частицы, так как они могут поглощать некоторую долю возбуждающей энергии, дезактивировать возбужденные молекулы или завышать измеряемую величину из-за многократных отражений в кювете с образцом.
Интенсивность флуоресценции обратно пропорциональна концентрации кислорода, являющегося сильным гасителем флуоресценции. По степени тушения флуоресценции можно определять концентрацию кислорода в окружающей среде. Для удаления кислорода через испытуемый образец пропускают азот или гелий.
Большинство флуоресцирующих веществ чувствительно к свету. Во время облучения во флуориметре они могут подвергаться фоторазложению с образованием других флуоресцирующих продуктов. Такие эффекты обнаруживаются при наблюдении за откликом детектора во времени и могут быть снижены путём приглушения света с помощью светофильтров или экранов.
Применение флуориметрии в фармацевтическом анализе
Идентификация. Спектры флуоресценции специфичны для определяемых веществ. Поэтому флуоресценция может быть применена для их идентификации.
Количественный анализ. При количественных определениях интенсивность флуоресценции раствора испытуемого образца сравнивают с интенсивностью флуоресценции раствора стандартного образца флуоресцирующего вещества известной концентрации, измеренной в идентичных условиях на одном и том же приборе.
Методика. Растворяют испытуемый образец в растворителе или в смеси растворителей, указанных в нормативной документации. Переносят раствор в кювету или пробирку флуориметра и облучают возбуждающим светом при длине волны, указанной в нормативной документации.
Измеряют интенсивность испускаемого света под углом 90 о к возбуждающему свету после прохождения через светофильтр или монохроматор, пропускающий преимущественно испускаемый диапазон длин волн.
Последовательность выполнения анализа. Вначале в прибор помещают растворитель или смесь растворителей, используемых для растворения вещества, и устанавливают регистрирующее устройство на нулевое значение. Затем вводят раствор стандартного образца и устанавливают чувствительность прибора таким образом, чтобы отклик показаний был не менее 50. Если для регулировки чувствительности требуется изменение ширины щели, должны быть повторены обнуление прибора на растворитель и измерение интенсивности флуоресценции стандартного образца. После этого вводят растворы испытуемых образцов неизвестной концентрации и регистрируют показания прибора. В случае линейной зависимости интенсивности испускаемого света от концентрации вещества рассчитывают последнюю в испытуемом растворе (C) по формуле:
где C0 — концентрация вещества в стандартном растворе;
I — интенсивность света, испускаемого испытуемым раствором;
I0 — интенсивность света, испускаемого стандартным раствором.
Если интенсивность флуоресценции не прямо пропорциональна концентрации раствора, измерение может быть произведено с использованием калибровочной кривой.
В некоторых случаях измерение флуоресценции испытуемого образца может быть выполнено относительно независимого стандарта (например, флуоресцентного стекла или раствора другого флуоресцентного вещества). В качестве стандартов могут быть использованы: раствор известной концентрации хинина в 0,05 М растворе серной кислоты или раствор флуоресцеина в 0,1 М растворе натрия гидроксида. В таких случаях концентрацию испытуемого образца следует определять с использованием предварительно полученной в тех же условиях калибровочной кривой.
Глава 2. Экспериментальная часть
2.1 Определение содержания рибофлавина (витамина В2) методом флуориметрии
Метод основан на сравнение интенсивности флуоресценции стандартного и анализируемого растворов витамина В1 при облучении светом ультрафиолетового диапазона. Содержание витамина В1 определяют, не прибегая к построению градуировочного графика лишь по одной точке, стандартный раствор при этом готовят с содержанием рибофлавина, близким к определяемому содержанию и не превышающим 1. 10-5 моль/дмЗ.
Необходимыми реактивами являются рабочий стандартный раствор и анализируемый раствор.
Для приготовления рабочего стандартного раствора, содержащего 0,04 мг/смЗ рибофлавина, взвешивают 0,0400 г рибофлавина в стаканчике или в фарфоровой чашке (навеску пересчитывают на чистую субстанцию). Затем навеску растворяют в горячей воде и переносят в мерную колбу объемом 1 дмЗ на водяной бане. После охлаждения объем доводят до метки дистиллированной водой. Раствор годен в течение 1 месяца при условии его хранения в бутылке из темного стекла в холодильнике при температуре от 5 до 10 ОС. В день проведения испытаний часть основного раствора переносят в стаканчик и выдерживают в темном месте до приобретения комнатной температуры, затем отбирают пипеткой 10 смЗ раствора в мерную колбу объемом 100 смЗ и доводят объем раствора до метки (1 смЗ раствора содержит 0,0004 мг/смЗ рибофлавина). Раствор годен только в день приготовления, а между измерениями хранится в темном месте.
В качестве анализируемого раствора используют или готовый раствор рибофлавина, или его готовят следующим образом: навеску растертых в ступке драже массой около 1 г взвешивают с точностью до 0,0002 г, растворяют в горячей дистиллированной воде при подогревании на водяной бане и количественно переносят в мерную колбу объемом 500 смЗ. Затем раствор охлаждают, доводят объем до метки и фильтруют через обычную фильтровальную бумагу (первые 10 смЗ отбрасывают). Из полученного раствора готовят раствор второго разведения. Для этого отбирают 10 смЗ раствора в мерную колбу на 100 смЗ и доводят до метки дистиллированной водой. Расчетное содержание рибофлавина в растворе около 0,4 мкг/смЗ или 0,0004 мг/смЗ. Между замерами раствор хранится в темном месте. [10, 29]
Определение содержания рибофлавина проводят на приборе «Квант», внешний вид которого представлен на рисунке:
Внешний вид флуориметра «Квант»
1- включение прибора в сеть; 2 — чувствительность; 3 — измерение;
4- переключатель установка 100 % грубо; 5 — установка 0 %;
6 — установка 100 % точно; 7 — отсчетный лимб;
8 — шкала отсчетного лимба; 9 — первичный фильтр; 10 — вторичный светофильтр; 11 — кюветное отделение; 12 — установка нуля;
13 — установка чувствительности; 14 — миллиамперметр.
Для проведения измерений на флуориметре «Квант» необходимо соблюдать следующий порядок:
1. Включить прибор в сеть переменного тока с напряжением 220 В и нажать кнопку 1 «сеть», при этом загорается индикаторная лампочка.
2. Дать прибору прогреться не менее 60 мин.
3. По спектрам поглощения и флуоресценции (экспериментальные или литературные данные) выбрать необходимые первичный и вторичный светофильтры. При установке светофильтра необходимо следить, чтобы зеркальная поверхность первичного светофильтра была обращена к источнику света, а вторичного — к кювете.
При определении рибофлавина первый светофильтр синий (длина волны 440 нм), а второй светофильтр — зеленый (длина волны 520 нм).
4. Установить электрический нуль прибора, проделав следующие операции:
-перевести в левое положение ручку «0 %» до упора;
-вращением ручки «установка нуля» установить стрелку индикатора миллиамперметра на нуль.
5. Определить фон «холостого опыта» или растворителя и провести его компенсацию:
-установить в прибор кювету с растворителем («холостая проба»);
-нажать кнопку «Измерение» и вращением ручки отсчетного лимба установить стрелку индикатора на нуль (показания лимба будут соответствовать величине фона растворителя («холостого опыта»));
-установить на нуль шкалу отсчетного лимба «%», нажать кнопку «Измерение» и вращением ручки «установка 0 %» вывести стрелку индикатора на нуль.
Фон растворителя будет скомпенсирован, и при дальнейших измерениях его можно не учитывать.
Внимание! При определении содержания рибофлавина «холостой пробой» является дистиллированная вода.
6. Заполнить кювету стандартным раствором. Нажать кнопку «Чувствительность» и вращением ручки «Установка чувствительности» добиться отклонения стрелки индикатора до деления 20 слева от 0. При этом надо следить, чтобы был выставлен 0 % на отсчетном лимбе и ручка «0 %» находилась в левом положении.
7. Не вынимая кювету с эталонным раствором, установить шкалу отсчетно- го лимба на деление 50 %. Нажать кнопку 1 переключателя «грубо», кнопку «Измерение» и вращением ручки «установка 100 % точно» установить стрелку индикатора на нуль. Если стрелка на нуль не устанавливается, включить кнопку
2 или 3 на переключателе «грубо», а затем нажать кнопку «Измерение» и ручкой «установка 100 % точно» добиться установки стрелки индикатора на нуль.
8. Определить интенсивность флуоресценции анализируемого раствора:
-установить в прибор кювету с анализируемым раствором;
-нажать на клавишу «Измерение» и вращением отсчетного лимба установить стрелку индикатора на нуль. Показания лимба будут соответствовать интенсивности светового потока флуоресценции исследуемого раствора в процентах.
Обработка результатов анализа
Содержание рибофлавина в одном драже (С) в граммах вычисляют по формуле
где А — показания флуориметра при замере испытуемого раствора;
В — показания флуориметра при замере стандартного образца;
А1 и В1 — показания флуориметра после гашения флуоресценции гидросульфитом натрия в растворах ( из опытных данных равно 1).
V1— объем испытуемого раствора, взятый для разведения (10 см 3 );
V2 — окончательный объем разведения (100 см 3 );
500 — первичный объем раствора, см 3 ;
а — масса навески, взятая для испытания, г;
б— средняя масса одного драже, г;
1000 — коэффициент пересчета в граммы.
Содержание рибофлавина в готовом растворе (С, мг/см 3 ) вычисляют по формуле
где Сст — содержание рибофлавина в стандартном растворе, мг/см 3 .
2.2 Статистический расчёт содержания рибофлавина (витамина В2) методом флуориметрии
Источник