Исследование «Бификардио» на экспериментальных животных
ДОКЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ «БИФИКАРДИО»

«Бификардио» в эксперименте с крысами линии Вистар
На базе биотехнологического центра ВСГУТУ была изучена эффективность БАД «Бификардио» на экспериментальных животных.
Пробиотическая биологически активная добавка к пище «Бификардо» как и все перспективные препараты, предназначающиеся для использования в практическом здравоохранении, прошла доклинические исследования путем проведения опытов над экпериментальными животными, в частности, над крысами.
Цель исследования – Изучить влияние БАД «Бификардио» на основные показатели липидного обмена экспериментальных животных.
Исследование проводилось в соответствии с МУК 2.3.2.721-98.2.3.2 (Институт питания РАМН). Объектом исследования стали половозрелые крысы линии Вистар (масса – 180-220 г). Алиментарную дислипидемию формировали с помощью атерогенной диеты путем внесения корм холестерина в количестве 5% от количества корма (0,25 г) в течении 14 суток.
Интактная группа – крысы, находившиеся на стандартном рационе вивария; опытные животные, содержавшиеся на экспериментальном рационе: опытная (гр. 1) на атерогенном рационе; опытная (гр.2) с использованием БАД «Бификардио» с льняным маслом; опытная (гр. 3) — с использованием БАД «Бификардио» с кедровым маслом на фоне атерогенной диеты.
Для коррекции липидов крови использовали микробную биомассу БАД «Бификардио», полученную путем культивирования специально подобранного штамма бифидобактерий Bifidobacterium longum DK-100 на питательной среде на основе молочной сыворотки с добавлением кедрового или льняного масла. Однократный объем вводимых БАД «Бификардио» в среднем составлял 3 мл/кг массы крысы.
Эвтаназию животных осуществляли путем декапитации под эфирным наркозом в соответствии с требованиями Европейской конвенции по защите экспериментальных животных 86/609 ЕЕС.
 Материалом исследования являлась кровь, взятая утром до кормления из шейной вены после декапитации крыс. Основные показатели липидного обмена в сыворотке крови определяли на автоматическом биохимическом анализаторе BS-400 (KHP).
Материалом исследования являлась кровь, взятая утром до кормления из шейной вены после декапитации крыс. Основные показатели липидного обмена в сыворотке крови определяли на автоматическом биохимическом анализаторе BS-400 (KHP).
Статистическую значимость различий средних величин оценивали с помощью t-критериев Стьюдента, достоверными считались результаты при p
Изучение показателей липидного обмена показало, что у крыс под влиянием атерогенного рациона (опыт 1) через 14 суток наблюдалось повышение ХС (на 36%), ТГ (на 40%), ЛПНП (на 30%) в сыворотке крови (табл.). При этом содержание ЛПВП снизилось на 25%, а Индекс Атерогенности повысился три раза (табл.).
Таблица – Влияние «Бификардио» на основные показатели липидного обмена животных
Источник
Регуляторные и методические аспекты доклинических и клинических исследований кормовых добавок для животных
Д.Р. Каргопольцева(1), ветеринарный врач группы специфической токсикологии, К.Л. Крышень(1), кандидат биологических наук, руководитель отдела токсикологии и микробиологии, М.Н. Макарова(2), доктор медицинских наук, директор, В.Г. Макаров(1), доктор медицинских наук, заместитель директора НПО «Дом Фармации» 1-Институт доклинических исследований, 188663, Россия, Ленинградская обл., Всеволожский район, г.п. Кузьмоловский, ул. Заводская, д. 3, к. 245; 2-Научно-производственное объединение «Дом Фармации», 188663, Россия, Ленинградская обл., Всеволожский район, г.п. Кузьмоловский, ул. Заводская, д. 3, к. 245 Е-mail: agasieva.dr@doclinika.ru
Резюме
В последние годы в связи с развитием сельского хозяйства и ростом числа владельцев домашних животных происходит стремительное развитие ветеринарии не только в Российской Федерации, но и во всем мире. Попутно с развитием ветеринарной медицины идет и совершенствование ветеринарной фармацевтики, которая разрабатывает и производит все больше ветеринарных препаратов и кормовых добавок для животных. Для попадания на рынок новых кормовых добавок для животных, так же как и лекарственных препаратов для человека, требуется государственная регистрация с соответствующим пакетом документов. Наиболее важные документы – результаты доклинических и клинических исследований. Однако если в отношении лекарственных препаратов для человека в Российской Федерации существует множество нормативных документов, которые описывают объем и схему проведения исследований, то в отношении кормовых добавок таковых документов и инструкций до настоящего времени не разработано. Поэтому требуется разработка регламентирующих документов, гармонизированных с международными требованиями. В данной работе рассмотрены основные международные регуляторные требования к проведению доклинических и клинических исследований кормовых добавок для животных.
Введение
В последние годы в Российской Федерации особое внимание уделяется развитию и совершенствованию сельского хозяйства. Сельское хозяйство – одна из ключевых и фундаментальных отраслей экономики любого государства, направленная на обеспечение населения продуктами питания и получение сырья для ряда областей промышленности. Одной из отраслей сельского хозяйства является животноводство, успех развития которого зависит от здоровья и продуктивности сельскохозяйственных животных. В связи с этим наблюдается непрерывное расширение ассортимента кормовых добавок для животных, которые не всегда отличаются хорошим качеством.
В п. 4 проекта Административного регламента предоставления Россель-хознадзором государственной услуги по государственной регистрации кормовых добавок приведено следующее определение: «кормовые добавки – это продукты или их комбинации растительного, животного, микробиологического, минерального и синтетического происхождения, предназначенные для включения в состав кормов и рационов животных с целью обеспечения физиологической полноценности, стимуляции продуктивности животных, обеспечения сохранности компонентов, увеличения доступности питательных веществ, улучшения вкусовых и технологических свойств кормов» [1].
Использование некачественных кормовых добавок может негативно сказаться не только на самих сельскохозяйственных животных, но и на здоровье людей, потребляющих продукцию, получаемую от этих животных.
Для поступления кормовой добавки в ветеринарную аптеку необходимо пройти государственную регистрацию, которую осуществляет Россельхознадзор на базе государственного учреждения «Всероссийский государственный центр контроля качества и стандартизации лекарственных средств для животных и кормов» (ФГУ «ВГНКИ») в течение 6 мес со дня подачи регистрационных документов. В соответствии с приказом Минсельхоза РФ №48 от 1 апреля 2005 г. «Об утверждении Правил государственной регистрации лекарственных средств для животных и кормовых добавок» [2] обязательной регистрации подлежат:
- новые добавки;
- новые комбинации зарегистрированных ранее добавок;
- добавки, зарегистрированные ранее, но произведенные в других формах, или с новой дозировкой, или с другим составом вспомогательных веществ;
- воспроизведенные добавки.
Для государственной регистрации кормовых добавок в Россельхознадзор необходимо предоставлять результаты доклинических исследований. Проведение доклинических исследований является обязательной процедурой для регистрации кормовой добавки и занесения ее в Государственный реестр кормовых добавок.
Таким образом, производитель должен составить программу и провести доклинические исследования разрабатываемых кормовых добавок для животных с целью изучения их эффективности, безопасности применения для здоровья животных и подтверждения их качества.
Как и в каком объеме проводить доклинические исследования кормовых добавок для животных? Для медицинских препаратов давно существуют руководства, стандартные правила и методические указания по организации и проведению доклинических исследований. В отношении ветеринарных препаратов до недавнего времени такие правила отсутствовали. Сегодня разработан Приказ Министерства сельского хозяйства Российской Федерации №101 от 06.03.2018 «Об утверждении правил проведения доклинического исследования лекарственного средства для ветеринарного применения, клинического исследования лекарственного препарата для ветеринарного применения, исследования биоэквивалентности лекарственного препарата для ветеринарного применения» [3]. В то же время в отношении кормовых добавок для животных правил по проведению исследований до настоящего времени в Российской Федерации не разработано. В связи с этим целью данной публикации стало рассмотрение международных регламентирующих документов.
Международные регуляторные требования
При рассмотрении вопроса о кормовых добавках в Европейских странах руководствуются Регламентом комиссии (ЕС) №1831/2003 [4], который описывает порядок выдачи разрешений на размещение на рынке и применение кормовых добавок, правил контроля и маркировки кормовых добавок и премиксов в качестве основы для обеспечения высокого уровня защиты здоровья человека, здоровья и благополучия животных, окружающей среды и интересов потребителей, а также обеспечение эффективного функционирования внутреннего рынка. При проведении исследований токсичности и эффективности кормовых добавок руководствуются Регламентом №429/2008 [5], в котором содержатся правила по подготовке и представлению заявок, а также проведению оценки и выдаче разрешений в отношении кормовых добавок.
В соответствии с Регламентом (ЕС) №1831/2003 [4], если дело касается кормовых добавок, то исследования токсичности и эффективности не требуются в отношении:
- мочевины;
- аминокислот, их солей и аналогов;
- микроэлементов и витаминов, провитаминов и веществ с установленным химическим составом, имеющих схожее действие и не накапливающихся в организме, уже разрешенных к применению в виде кормовых добавок;
- кормовых добавок, произведенных путем ферментации, в случае если производственный организм рассматривается европейским агентством по безопасности пищевых продуктов в качестве организма с установленным безопасным статусом;
- кормовых добавок, произведенных путем ферментации, действующее вещество которых отделяется от необработанного ферментированного продукта и подвергается высокой очистке.
Токсикологические исследования и исследования эффективности проводятся в целях совершенствования состава:
- витаминов, провитаминов и веществ с установленным химическим составом, имеющих схожее действие и способных к накоплению, предполагаемая или подтвержденная способность к накоплению которых отличается от способности к накоплению витамина(-ов) с установленным составом;
- производных мочевины;
- аналогов аминокислот и соединений микроэлементов ранее не одобренных;
- кормовых добавок, произведенных путем ферментации, не исключенных по вышеописанным правилам.
Исследования токсичности и эффективности кормовых добавок для животных проводят на лабораторных животных (проявляющих наибольшую чувствительность) и на целевых видах животных (табл. 1). Такие исследования осуществляют в соответствии с Директивой 2010/63/EU Европейского Парламента и Совета Европейского Союза по охране животных, используемых в научных целях, от 22.09.2010 [6].
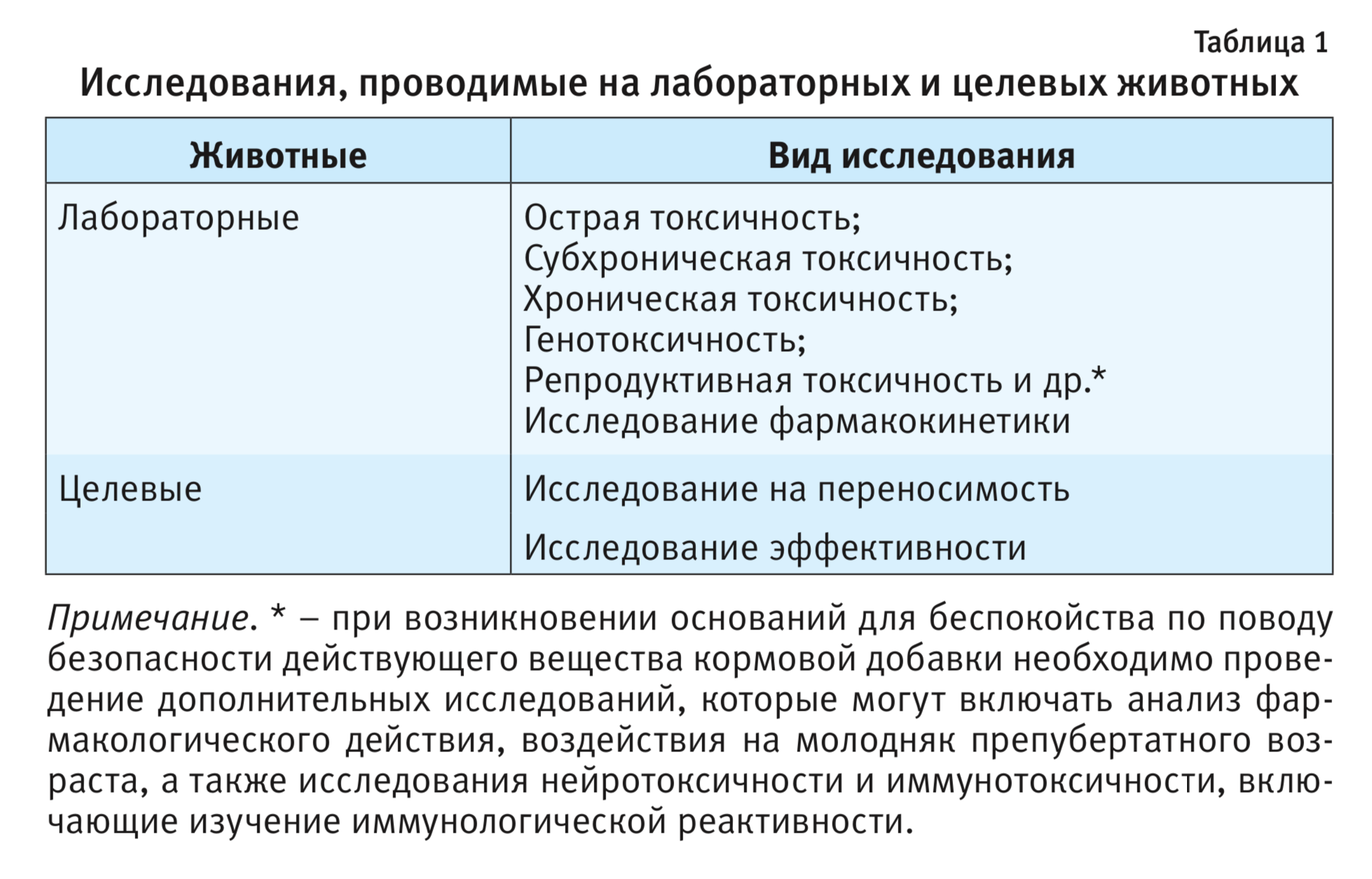
Далее рассмотрим необходимые исследования, проводимые на лабораторных и целевых животных.
Доклинические исследования кормовых добавок для животных, проводимые на лабораторных животных
Изучение острой токсичности
Исследования острой токсичности проводятся с целью установления принадлежности тестируемого объекта к определенному классу токсичности. Исследования острой токсичности осуществляются не менее чем на 2 видах млекопитающих. В случае необходимости один вид лабораторных животных может быть заменен целевым видом. Точное определение летальной дозы LD50 не является необходимостью, достаточным считается определение приблизительного значения летальной дозы [4, 5]. При проведении токсикологических исследований по изучению острой токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.420 «Acute Oral Toxicity – Fixed Dose Procedure», ГОСТ 32296-2013; Test No.423 «Acute Oral Toxicity – Acute Toxic Class Method», ГОСТ 32644-2014) [7].
Изучение субхронической токсичности
Изучение субхронической токсичности тестируемых объектов проводят на мышах и крысах при пероральном/внутрижелудочном введении в течение не менее 90 сут. При необходимости исследование субхронической токсичности дополнительно проводится на животных, не относящихся к отряду грызунов. Для установления дозозависимых эффектов тестируемых объектов в исследование субхронической токсичности должно быть включено не менее 3 уровней доз, при этом минимальная доза не должна оказывать токсического действия на организм. Также должна быть сформирована контрольная группа, с помощью которой оцениваются результаты проведенного экспериментального исследования [4, 5]. При проведении исследований по изучению субхронической токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.408 «Repeated Dose 90-Day Oral Toxicity Study in Rodents», ГОСТ 32637-2014; Test No.409 «Repeated Dose 90-Day Oral Toxicity Study in Non-Rodents», ГОСТ Р 56697-2015) [7].
Изучение хронической токсичности
Для установления хронической токсичности исследования тестируемых объектов выполняют на протяжении не менее 12 мес. Так же, как при изучении субхронической токсичности, изучение хронической токсичности происходит с исследованием как минимум 3 доз и с добавлением контрольной группы животных [4, 5]. При проведении исследований по изучению хронической токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.452 «Chronic Toxicity Studies», ГОСТ 32437-2013; Test No.453 «Combined Chronic Toxicity/Carcinogenicity Studies», ГОСТ 32647-2014) [7].
В документе Регламента комиссии (ЕС) № 429/2008 не предусмотрено более подробное рассмотрение вопроса о длительности проведения исследования хронической токсичности, а также возможности проведения исследования хронической токсичности без исследования субхронической токсичности. Такие длительные исследования очень дорогостоящие и трудозатратные. Заметим, что доклинические исследования лекарственных препаратов для человека регламентируются ГОСТ Р 56701-2015 [8], где рекомендуемая продолжительность исследования общетоксических свойств при многократном применении у животных соответствует продолжительности применения препарата в клинике (табл. 2).
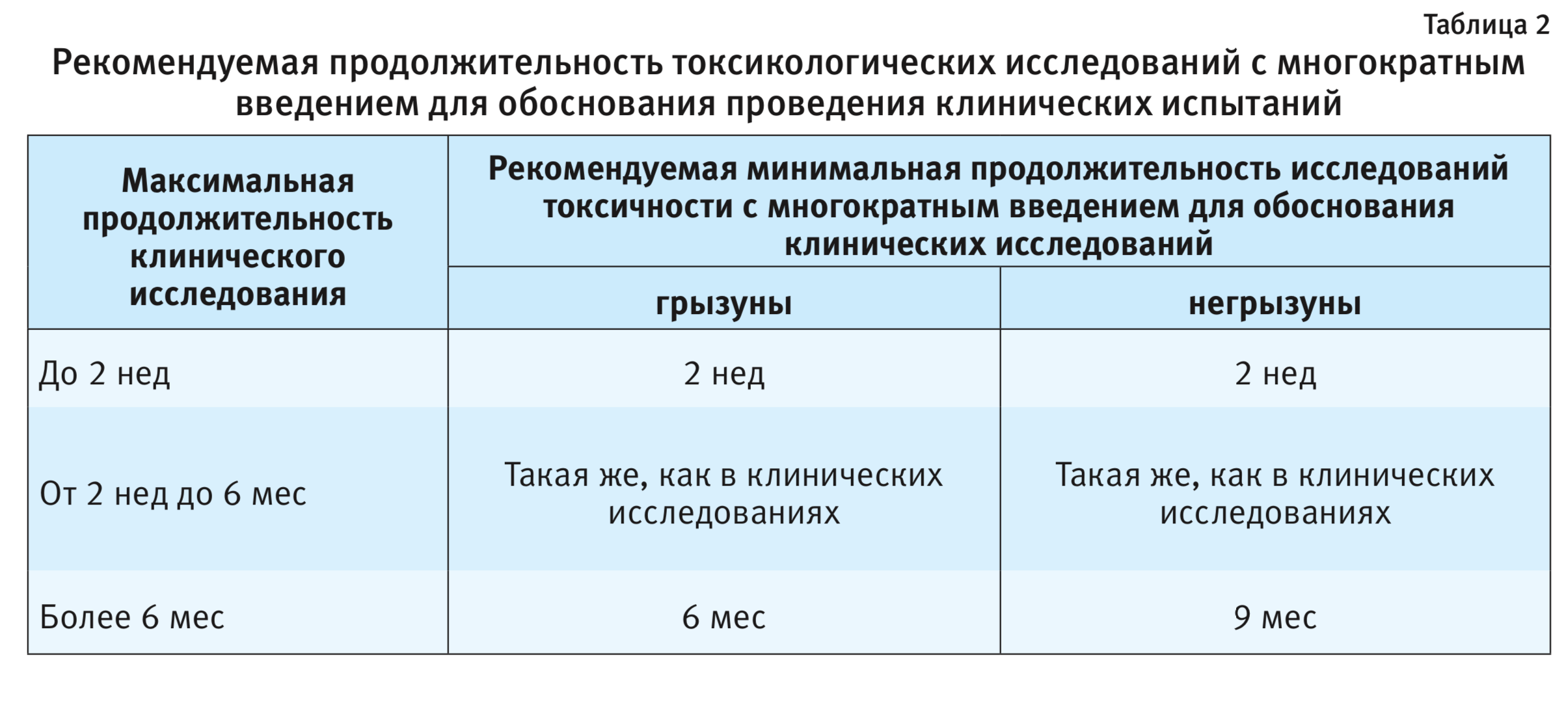
Изучение генотоксичности
Для изучения генотоксичности тестируемых объектов существует целый ряд испытаний:
- тесты на индукцию генных мутаций у бактерий и в клетках млекопитающих;
- индукция хромосомных аберраций в клетках млекопитающих;
- испытания in vivo.
При проведении исследований по изучению генотоксичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.471 «Bacterial Reverse Mutation Test», ГОСТ 32376-2013, Test No.473 «In Vitro Mammalian Chromosomal Aberration Test»; Test No.474 «Mammalian Erythrocyte Micronucleus Test» ГОСТ 32635-2014; Test No.476 «Mammalian Bone Marrow Chromosomal Aberration Test»; Test No.476 «In Vitro Mammalian Cell Gene Mutation Tests using the Hprt and xprt
genes») [7].
В регламенте Комиссии (ЕС) №429/2008 нет четких рекомендаций по объему исследований генотоксичности кормовых добавок для животных. Если рассматривать исследования генотоксичности лекарственных препаратов для человека, то существуют рекомендации ГОСТ 57130-2016 [9], где предлагается проведение следующей батареи тестов:
Вариант 1
- Испытание генных мутаций в бактериях.
- Цитогенетический тест хромосомных повреждений (анализ хромосомных аберраций в метафазе invitro или микроядерная проба in vitro) либо анализ генных мутаций Tk в лимфоме мышей in vitro.
- Тест генотоксичности invivo, обычно тест на хромосомные поломки на гемопоэтических клетках мышей в виде либо микроядерной пробы либо анализа на хромосомные аберрации в метафазе.
Вариант 2
- Испытание генных мутаций в бактериях.
- Оценка генотоксичности invivo двух разных тканей, обычно микроядерная проба с использованием гемопоэтических клеток грызунов и 2-й анализ in vivo. Как правило, используется анализ поломки нитей ДНК в печени.
Изучение репродуктивной токсичности
Цель изучения репродуктивной токсичности – установление возможных неблагоприятных воздействий тестируемого объекта на мужскую и женскую репродуктивную систему, а также патологического воздействия на развитие потомства. При проведении исследований репродуктивной токсичности тестируемый объект вводят в течение определенного времени до ссадки самцов и самок. Затем наблюдают за эффективностью оплодотворения, течением беременности и родов у самок, далее контролируют рост и развитие потомства [4, 5]. При проведении исследований по изучению репродуктивной токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.416 «Two-Generation Reproduction Toxicity Study», ГОСТ Р 56698-2015) [7].
Также изучение репродуктивной токсичности включает в себя исследование токсических эффектов на внутриутробное развитие потомства, целью которого является установление неблагоприятного воздействия тестируемого объекта на беременных самок и плод при введении объекта на протяжении всего периода беременности [5, 6]. В ходе исследований следует соблюдать Руководящие принципы ОЭСР (OECD Test No.414 «Prenatal Development Toxicity Study», ГОСТ 32380-2013) [7].
В документе Регламента комиссии (ЕС) № 429/2008 также нет достаточно подробного описания необходимости проведения исследования тестируемых объектов на беременных самках. По нашему мнению, данной необходимости нет, если нет опасений в неблагоприятном воздействии кормовой добавки на течение беременности и плод в клинике и, если кормовая добавка не предусмотрена к применению в период беременности.
Исследование фармакокинетики кормовых добавок и остаточных веществ
Решающим фактором при идентификации и количественном определении остаточных веществ в продуктах питания, полученных от животных (молоко, мясо, субпродукты и др.), в рацион которых была включена кормовая добавка, является установление метаболического пути добавки у целевых видов животных. При исследовании фармакокинетики представляются результаты исследований всасывания, распределения, метаболизма и экскреции (выведения) действующего вещества (и его метаболитов) кормовой добавки.
Исследования фармакокинетики действующего вещества кормовых добавок должны проводиться при введении животным в максимальной дозе, согласно инструкции по применению кормовой добавки. Исследования предусматривают приблизительную оценку скорости и степени всасывания, распределения в крови и выделения с экскретами (с мочой, желчью, экскрементами, молоком, яйцами), а также установление фармакокинетического профиля, порядка распределения действующего вещества в тканях и продуктах после введения повторных доз до достижения метаболического равновесия.
Исследования должны включать в себя идентификацию остаточных веществ действующего вещества, оказывающих токсическое действие, в продуктах животноводства (печень, почки, мышцы, кожа, кожа + жир, молоко, яйца), а также установление периода выведения.
Фармакокинетические исследования проводятся на лабораторных видах животных. Однако в случае исследований на определенные метаболиты могут потребоваться дополнительные исследования, если такие метаболиты вырабатываются в организме целевых животных и не образуются в значительной степени в организме лабораторных животных.
Исследования кормовых добавок для животных, проводимые на целевых животных
Согласно Регламентам (ЕС) №1831/2003 [4] и №429/2008 [5], исследования проводятся также на целевых видах животных, которые включают в себя выявление переносимости и эффективности кормовых добавок.
Исследование переносимости кормовых добавок
При установлении переносимости кормовой добавки проводят кратковременные исследования, длительность которых зависит от вида животных (табл. 3–7). Обычно исследуют максимально рекомендуемую дозу и дозы в 10 раз ее превышающие, при этом учитывают результаты доклинических исследований на лабораторных животных. В случае переносимости кормовых добавок целевыми видами животных в дозах, превышающих максимально рекомендуемые менее чем в 10 раз, при проведении исследования необходимо предусматривать определение предельно допустимого безопасного уровня добавки.
Источник